ธาลัสซีเมีย
Tanja Unterberger ศึกษาวารสารศาสตร์และวิทยาศาสตร์การสื่อสารในกรุงเวียนนา ในปี 2015 เธอเริ่มทำงานเป็นบรรณาธิการด้านการแพทย์ที่ ในออสเตรีย นอกจากการเขียนข้อความเฉพาะทาง บทความในนิตยสาร และข่าวแล้ว นักข่าวยังมีประสบการณ์ในด้านพอดแคสต์และการผลิตวิดีโออีกด้วย
ข้อมูลเพิ่มเติมเกี่ยวกับผู้เชี่ยวชาญของ เนื้อหา ทั้งหมดได้รับการตรวจสอบโดยนักข่าวทางการแพทย์ธาลัสซีเมียหรือโรคโลหิตจางในทะเลเมดิเตอร์เรเนียนเป็นโรคทางพันธุกรรมของเซลล์เม็ดเลือดแดง ยีนที่บกพร่องทำให้ร่างกายผลิตเม็ดเลือดแดง (ฮีโมโกลบิน) น้อยเกินไป หรือถูกทำลายลงเร็วเกินไป ขึ้นอยู่กับตำแหน่งของข้อบกพร่องทางพันธุกรรม ความแตกต่างระหว่างอัลฟาและเบตาธาลัสซีเมีย ทั้งสองรูปแบบนำไปสู่โรคโลหิตจาง (โรคโลหิตจาง) แพทย์มักจะรักษาด้วยยาและการถ่ายเลือด อ่านเพิ่มเติมเกี่ยวกับสาเหตุ อาการ การวินิจฉัย และการรักษาที่นี่!
รหัส ICD สำหรับโรคนี้: รหัส ICD เป็นรหัสที่เป็นที่ยอมรับในระดับสากลสำหรับการวินิจฉัยทางการแพทย์ สามารถพบได้เช่นในจดหมายของแพทย์หรือในใบรับรองความสามารถในการทำงาน D57D56

ภาพรวมโดยย่อ
- คำอธิบาย : โรคที่กำหนดทางพันธุกรรมของเซลล์เม็ดเลือดแดง (เม็ดเลือดแดง) ที่นำไปสู่โรคโลหิตจาง
- การวินิจฉัย: แพทย์วินิจฉัยโรคธาลัสซีเมียโดยการตรวจเลือดแบบพิเศษและการวิเคราะห์สารพันธุกรรม (การวิเคราะห์ดีเอ็นเอ)
- สาเหตุ: ความบกพร่องทางพันธุกรรมที่สืบทอดมาซึ่งทำให้ร่างกายผลิตเม็ดเลือดแดง (ฮีโมโกลบิน) น้อยเกินไปหรือไม่มีเลย
- การรักษา: การรักษาคือการใช้ยาและการถ่ายเลือด ในบางกรณีจำเป็นต้องปลูกถ่ายไขกระดูก
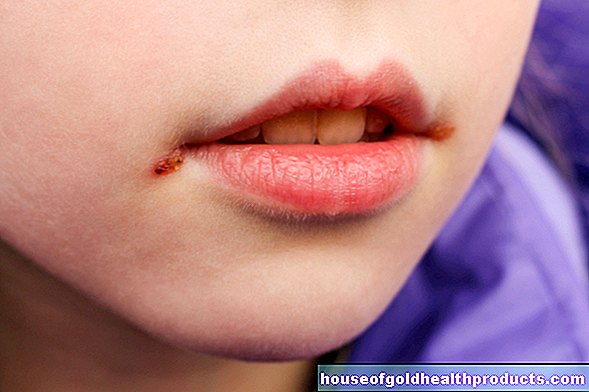
- อาการ: เหนือสิ่งอื่นใด, โรคโลหิตจาง, อ่อนเพลีย, ตับและม้ามโต, ความผิดปกติของการเจริญเติบโต, การเปลี่ยนแปลงของกระดูก, โรคกระดูกพรุน; ธาลัสซีเมียที่ไม่รุนแรงมักไม่มีอาการ
- การพยากรณ์โรค: ธาลัสซีเมียมีระดับความรุนแรงต่างกัน ยิ่งรักษาอาการเร็ว การพยากรณ์โรคก็จะยิ่งดีขึ้น ปัจจุบันการรักษาทำได้ด้วยการปลูกถ่ายเซลล์ต้นกำเนิดหรือการบำบัดด้วยยีนเท่านั้น
ธาลัสซีเมียคืออะไร?
ธาลัสซีเมีย (เช่นโรคโลหิตจางในทะเลเมดิเตอร์เรเนียน) เป็นกลุ่มของโรคทางพันธุกรรมที่การก่อตัวของเซลล์เม็ดเลือดแดง (เม็ดเลือดแดง) ถูกรบกวนโดยข้อบกพร่องทางพันธุกรรม เป็นผลให้ร่างกายผลิตเม็ดเลือดแดง (ฮีโมโกลบิน) น้อยเกินไปหรือไม่มีเลยหรือสลายตัวเร็วเกินไป
ผลจากการเปลี่ยนแปลงของฮีโมโกลบิน เซลล์เม็ดเลือดมักจะมีขนาดเล็กกว่าปกติและมีอายุขัยสั้นลง ซึ่งนำไปสู่ภาวะโลหิตจางเมื่อเวลาผ่านไป พร้อมด้วยอาการทั่วไป เช่น เหนื่อยล้าและหัวใจเต้นเร็ว
ธาลัสซีเมียเป็นโรคเลือดที่พบได้บ่อยในเด็กและวัยรุ่น พวกเขายังอยู่ในกลุ่มของ hemoglobinopathies (ความผิดปกติของฮีโมโกลบิน) โรคธาลัสซีเมียสามารถถ่ายทอดจากรุ่นสู่รุ่นได้ เนื่องจากธาลัสซีเมียแพร่หลายมากในภูมิภาคเมดิเตอร์เรเนียน จึงเรียกอีกอย่างว่า "โรคโลหิตจางในเมดิเตอร์เรเนียน"
ธาลัสซีเมียพัฒนาได้อย่างไร?
เฮโมโกลบินเป็นโปรตีนที่พบในเซลล์เม็ดเลือดแดงและสร้างขึ้นในไขกระดูก ช่วยให้เซลล์เม็ดเลือดแดงนำออกซิเจนที่สำคัญจากปอดไปยังทุกส่วนของร่างกาย
โดยทั่วไปแล้ว เฮโมโกลบินประกอบด้วยสายโปรตีนสี่สาย โดยสองสายมีความเหมือนกัน - สองสายอัลฟาและสายเบตาสองสาย เฮโมโกลบินยังมีธาตุเหล็กซึ่งจับออกซิเจน ในโรคธาลัสซีเมีย ร่างกายจะสร้างสายโปรตีนน้อยเกินไปหรือมีการเปลี่ยนแปลง (สายอัลฟาหรือเบตา) เนื่องจากยีนที่เปลี่ยนแปลง (การกลายพันธุ์)
ซึ่งหมายความว่าสามารถสร้างโมเลกุลของเฮโมโกลบินที่ใช้งานได้น้อยลง เซลล์เม็ดเลือดแดงหดตัวและสลายตัวมากขึ้นเรื่อยๆ สิ่งนี้นำไปสู่ภาวะโลหิตจาง เนื่องจากร่างกายไม่ได้รับออกซิเจนอย่างเพียงพออีกต่อไปเนื่องจากขาดเซลล์เม็ดเลือดแดง
ขึ้นอยู่กับห่วงโซ่โปรตีน (สายอัลฟาหรือเบต้า) ที่ได้รับผลกระทบ ความแตกต่างระหว่าง α- (อัลฟา) -ธาลัสซีเมีย และ β- (เบต้า) -ธาลัสซีเมีย
อัลฟ่าธาลัสซีเมีย
รูปแบบที่พบได้น้อยกว่าคือ α-ธาลัสซีเมีย ในรูปแบบนี้ ร่างกายสร้างสายอัลฟาน้อยเกินไปหรือไม่มีเลย
เบต้าธาลัสซีเมีย
เบต้าธาลัสซีเมียเป็นรูปแบบที่พบบ่อยของธาลัสซีเมีย ในรูปแบบนี้ ร่างกายสร้างสายเบต้าเฮโมโกลบินน้อยเกินไปหรือไม่มีเลย
มีธาลัสซีเมียเดลต้าและแกมมาด้วย แต่ทั้งคู่มีน้อยมากและมักไม่รุนแรง
การวินิจฉัยโรคธาลัสซีเมียเป็นอย่างไร?
เนื่องจากอาการของโรคธาลัสซีเมียมักปรากฏในวัยเด็ก จุดสัมผัสแรกจึงมักอยู่ที่กุมารแพทย์ หากจำเป็นและเพื่อการตรวจเพิ่มเติม เขาจะแนะนำคุณให้รู้จักกับผู้เชี่ยวชาญด้านอายุรศาสตร์ (นักโลหิตวิทยา)
ประวัติครอบครัว
เนื่องจากธาลัสซีเมียเป็นโรคที่สืบทอดมา ประวัติครอบครัวมักจะให้ข้อมูลเบื้องต้นแก่แพทย์ ตัวอย่างเช่น แพทย์ถามว่าในครอบครัวมีพาหะนำโรคหรือไม่
การตรวจเลือด
เพื่อยืนยันการวินิจฉัย แพทย์จะทำการตรวจเลือดรวมทั้งเฮโมโกลบินอิเล็กโตรโฟรีซิส (Hb electrophoresis)
อิเล็กโตรโฟรีซิสของเฮโมโกลบิน
ด้วยเฮโมโกลบินอิเล็กโตรโฟรีซิส แพทย์จะละลายฮีโมโกลบินจากเลือดของผู้ป่วยในของเหลวและนำไปใช้กับวัสดุพาหะพิเศษ (เช่น ทำจากกระดาษหรือเจลาติน) จากนั้นเขาก็ใช้แรงดันไฟฟ้า
ขึ้นอยู่กับว่าฮีโมโกลบินประกอบขึ้นอย่างไร ฮีโมโกลบินจะเคลื่อนที่เป็นระยะทางต่างกันในช่วงเวลาที่กำหนด แพทย์จะประเมินว่าฮีโมโกลบินเป็นปกติหรือผิดปกติตามระยะทางที่ครอบคลุม
ภายใต้กล้องจุลทรรศน์ แพทย์มักจะตรวจพบเซลล์ที่มีรูปร่างลักษณะเฉพาะในเลือดของบุคคลนั้น เหล่านี้เป็นเซลล์เม็ดเลือดแดงที่ค่อนข้างซีดและมีฮีโมโกลบินสีเข้มอยู่ตรงกลาง เซลล์เม็ดเลือดมีขนาดเล็กและมีฮีโมโกลบินต่ำ
การวินิจฉัยในทารก
แพทย์แนะนำให้ทารกแรกเกิดที่เป็นธาลัสซีเมียในครอบครัวได้รับการตรวจเลือด (เช่น การตรวจคัดกรองทารกแรกเกิด) ในการทำเช่นนี้ แพทย์จะนำเลือดบางส่วน (เช่น จากสายสะดือ) ทันทีหลังคลอดและตรวจดูการเปลี่ยนแปลงทางพันธุกรรม วิธีนี้ทำให้สามารถรับรู้โรคได้ตั้งแต่เนิ่นๆ และรักษาได้ทันท่วงที
การวินิจฉัยขณะตั้งครรภ์
ในเด็กที่ยังไม่เกิด การตรวจธาลัสซีเมีย (การวินิจฉัยก่อนคลอด) จะดำเนินการโดยใช้ตัวอย่างเนื้อเยื่อจากรก
แพทย์แนะนำให้ผู้ปกครองที่ตั้งครรภ์ที่รู้ว่าตนเองเป็นพาหะธาลัสซีเมียให้ทำการวินิจฉัยก่อนคลอดในช่วงสิบสองสัปดาห์แรกของการตั้งครรภ์
ธาลัสซีเมียเป็นกรรมพันธุ์?
ธาลัสซีเมียเป็นกรรมพันธุ์ ซึ่งหมายความว่าผู้ที่เป็นโรคธาลัสซีเมียจะสืบทอดความบกพร่องทางพันธุกรรมจากพ่อแม่และเกิดมาพร้อมกับโรคนี้
มรดก: อัลฟาธาลัสซีเมีย
อัลฟ่าธาลัสซีเมียมีระดับความรุนแรงต่างกัน มีความแตกต่างระหว่าง alpha thalassemia minima, minor, intermedia และ major การแสดงออกขึ้นอยู่กับจำนวนของยีนทางพยาธิวิทยาที่พ่อแม่ส่งต่อไปยังเด็ก มีสี่ยีนสำหรับสายอัลฟา สองยีนจากแม่และอีกสองยีนจากบิดา
ยีนทั้งหมดสี่ยีนเกี่ยวข้องกับการก่อตัวของสายอัลฟา ในอัลฟาธาลัสซีเมียเมเจอร์ ยีนทั้งหมดที่เด็กสืบทอดมาจากพ่อแม่นั้นมีข้อบกพร่อง เป็นอัลฟาธาลัสซีเมียรูปแบบที่ร้ายแรงที่สุด เด็กที่มีรูปแบบนี้แทบจะไม่สามารถอยู่รอดและมักจะตายก่อนเกิดหรือสองสามวันหลังจากนั้น มีเพียงไม่กี่กรณีเท่านั้นที่สามารถทำให้เด็กๆ มีชีวิตอยู่ได้ด้วยความช่วยเหลือจากการปลูกถ่ายสเต็มเซลล์
ผู้ที่เป็นอัลฟาธาลัสซีเมียเล็กน้อยหรือคนกลางได้รับยีนบกพร่องสองหรือสามยีนจากพ่อแม่ พวกเขาอยู่ในรูปแบบที่รุนแรงกว่าของอัลฟาธาลัสซีเมีย และมักจะทำให้เกิดอาการในระดับปานกลาง ไม่รุนแรง หรือไม่มีเลยในผู้ที่ได้รับผลกระทบ
หากมีพ่อแม่เพียงคนเดียวที่สืบทอดยีนที่มีข้อบกพร่องให้ลูก เรียกว่า alpha thalassemia minima ผู้ที่ได้รับผลกระทบมักไม่มีอาการและมีชีวิตอยู่โดยปราศจากการด้อยค่า
การถ่ายทอดทางพันธุกรรม: เบต้าธาลัสซีเมีย
เบต้าธาลัสซีเมียก็มีระดับความรุนแรงแตกต่างกันไป พวกเขาจะแบ่งออกเป็นรายย่อย, สื่อกลางและที่สำคัญ
ธาลัสซีเมียเมเจอร์ (เรียกอีกอย่างว่าโรคโลหิตจางของคูลีย์) เป็นรูปแบบที่รุนแรงที่สุด มันเกิดขึ้นเมื่อพ่อแม่ทั้งสองส่งต่อความบกพร่องทางพันธุกรรมไปยังเด็ก
ในกรณีของ beta-thalassemia major จำเป็นต้องแลกเปลี่ยนเลือดของผู้ที่ได้รับผลกระทบอย่างสม่ำเสมอ (ทุกๆ 3 ถึง 4 สัปดาห์) โดยการให้เลือด หากปล่อยทิ้งไว้โดยไม่รักษา ผู้ที่ได้รับผลกระทบมักจะเกิดความผิดปกติของกระดูกอย่างรุนแรง เด็กที่ได้รับผลกระทบยังอ่อนแอต่อการติดเชื้อมากขึ้น พวกเขาอ่อนแอและมักจะไม่พัฒนาตามอายุ
เบต้าธาลัสซีเมียอินเตอร์มีเดียเป็นรูปแบบกลาง ผู้ที่มีรูปแบบนี้ยังได้รับยีนที่บกพร่องจากทั้งพ่อและแม่ อาการของพวกเขามักจะไม่เด่นชัดเท่าในรูปแบบที่สำคัญ คุณต้องการการถ่ายเลือดเป็นครั้งคราวเท่านั้น
ในเบต้าธาลัสซีเมียไมเนอร์ ผู้ที่ได้รับผลกระทบจะได้รับยีนลูกโซ่เบตาเฮโมโกลบินที่บกพร่องจากพ่อแม่เพียงคนเดียวเท่านั้น ผู้ปกครองอีกคนหนึ่งถ่ายทอดยีนที่ทำงานให้กับเด็ก
ผู้ที่เป็น beta-thalassemia minor มักไม่มีอาการโลหิตจางเล็กน้อย ตามกฎแล้วพวกเขาไม่ต้องการการรักษา อย่างไรก็ตาม ในฐานะที่เป็นพาหะของยีน พวกมันสามารถถ่ายทอดธาลัสซีเมียไปยังลูกๆ ได้
หากพ่อและแม่ทั้งสองมีโรคธาลัสซีเมียชนิดเบต้าเล็กน้อย มีโอกาสร้อยละ 25 ที่เด็กจะถ่ายทอดการกลายพันธุ์ทั้งสองแบบและพัฒนาเป็นธาลัสซีเมียที่สำคัญหรือปานกลาง มีโอกาส 50 เปอร์เซ็นต์ที่เด็กจะได้รับรูปแบบรองและโอกาส 25 เปอร์เซ็นต์ที่เด็กมีสุขภาพที่ดี
เพื่อที่จะได้รับเบต้าธาลัสซีเมีย ทั้งพ่อและแม่ต้องถ่ายทอดยีนที่บกพร่องไปให้เด็ก พาหะของโรคมียีนบกพร่องเพียงยีนเดียว อีกยีนหนึ่งทำงานได้ แม้ว่าพวกเขาจะไม่ป่วย แต่ก็สามารถถ่ายทอดยีนไปสู่ลูกหลานได้
ธาลัสซีเมียเกิดที่ไหน?
ธาลัสซีเมียยังเป็นที่รู้จักกันในนามว่าเป็นโรคโลหิตจางในทะเลเมดิเตอร์เรเนียน เนื่องจากเป็นที่แพร่หลายในประเทศแถบเมดิเตอร์เรเนียน เช่น อิตาลีและกรีซ แต่ยังเกิดขึ้นในใกล้และตะวันออกกลางและในส่วนของแอฟริกาและเอเชียในยุโรปกลาง โรคธาลัสซีเมียได้แพร่กระจายผ่านการเชื่อมโยงการค้าและการอพยพย้ายถิ่นทั่วโลก
ตั้งแต่ปี 1960 เป็นต้นมา โรคธาลัสซีเมียได้เกิดขึ้นในยุโรปตอนกลางและตอนเหนือเช่นกัน (เช่น เยอรมนี ออสเตรีย ฝรั่งเศส อังกฤษ เนเธอร์แลนด์ เบลเยียม และสแกนดิเนเวีย) ในระหว่างนี้ โรคธาลัสซีเมียเป็นโรคเลือดที่พบได้บ่อยในเด็กและวัยรุ่น
โรคนี้พบบ่อยแค่ไหน?
คาดว่าประมาณ 500 ถึง 600 คนในเยอรมนีอาศัยอยู่กับโรคธาลัสซีเมียแบบรุนแรง ประมาณ 200,000 คนในเยอรมนีป่วยด้วยโรคธาลัสซีเมียที่ไม่รุนแรง นอกจากนี้ เบต้าธาลัสซีเมียยังพบได้บ่อยกว่าอัลฟาธาลัสซีเมียอีกด้วย
รักษาโรคธาลัสซีเมียอย่างไร?
แพทย์ซึ่งมักจะเป็นผู้เชี่ยวชาญ (เช่น นักโลหิตวิทยาในเด็ก) จะรักษาธาลัสซีเมียโดยขึ้นอยู่กับความรุนแรงของโรคและอาการที่เกิดขึ้น การรักษาเกิดขึ้นในศูนย์ที่เชี่ยวชาญด้านโรค ทั้งนี้ขึ้นอยู่กับว่าผู้ที่ได้รับผลกระทบต้องการการถ่ายเลือด ปัจจุบันมีการแยกความแตกต่างระหว่างการรักษาธาลัสซีเมียที่ต้องอาศัยการถ่าย (TDT) กับการไม่ถ่ายเลือด (NTDT)
การรักษาแบบไม่พึ่งการถ่ายเลือด
ธาลัสซีเมียที่ไม่รุนแรงมักไม่ต้องการการรักษา อย่างไรก็ตาม ในบางกรณีที่พบไม่บ่อยนักในระหว่างตั้งครรภ์ สตรีมีครรภ์อาจมีภาวะโลหิตจางรุนแรงเนื่องจากธาลัสซีเมียที่ไม่รุนแรง จากนั้นพวกเขาต้องการการถ่ายเลือดเป็นประจำเพื่อปกป้องทั้งแม่และเด็ก
การรักษาขึ้นอยู่กับการถ่ายเลือด
แพทย์จะทำการแลกเปลี่ยนเลือดของผู้ที่ต้องอาศัยการถ่ายเลือด (เช่น กับธาลัสซีเมียเมเจอร์หรือสื่อกลาง) กับการถ่ายเลือดเป็นประจำ ขึ้นอยู่กับความรุนแรงของธาลัสซีเมีย ซึ่งมักเกิดขึ้นทุก ๆ วินาทีถึงสามสัปดาห์ โดยปกติตลอดชีวิต
การถ่ายเลือดเพิ่มเติมมักทำให้มีธาตุเหล็กมากเกินไป ในกรณีเหล่านี้ แพทย์จะสั่งยาเพื่อกำจัดธาตุเหล็กส่วนเกินออกจากร่างกาย (เรียกว่าคีเลเตอร์หรือธาตุเหล็ก) แพทย์จะฉีดเข้าไปใต้ผิวหนัง (ใต้ผิวหนัง) ที่โรงพยาบาลหรือผู้ที่เกี่ยวข้องรับประทานยาเม็ดที่บ้าน
การปลูกถ่ายเซลล์ต้นกำเนิด
เป็นไปได้ที่จะรักษาธาลัสซีเมียที่สำคัญด้วยการปลูกถ่ายสเต็มเซลล์ อย่างไรก็ตาม ถือว่ามีผู้บริจาคที่เหมาะสมกับบุคคลที่เกี่ยวข้อง
ยีนบำบัด
มีการบำบัดด้วยยีนสำหรับเบตาธาลัสซีเมียที่ต้องอาศัยการถ่ายเลือดมาตั้งแต่ปี 2019 การบำบัดรักษาความบกพร่องทางพันธุกรรมเชิงสาเหตุและให้ผู้ที่ได้รับผลกระทบมีโอกาสที่จะมีชีวิตอยู่โดยปราศจากการถ่ายเลือดไปตลอดชีวิต
ในการรักษาใหม่นี้ ยีนที่สมบูรณ์ซึ่งสามารถผลิตเฮโมโกลบินที่มีสุขภาพดีจะถูกฉีดเข้าไปในเซลล์ต้นกำเนิดในเลือดของบุคคลนั้น ในการทำเช่นนี้ เซลล์ต้นกำเนิดจะถูกนำออกจากไขกระดูกของผู้ป่วยก่อนแล้วจึงรักษาด้วยยา จากนั้นผู้ที่เกี่ยวข้องจะได้รับสเต็มเซลล์ของตัวเอง (ซึ่งมียีนที่ไม่บุบสลาย) กลับมา ซึ่งปัจจุบันสร้างเซลล์เม็ดเลือดแดงที่แข็งแรงและเม็ดเลือดปกติ
European Medicines Agency (EMA) ได้อนุมัติการรักษาสำหรับผู้ที่มีอายุสิบสองปีขึ้นไปซึ่งเหมาะสำหรับการรักษารูปแบบนี้โดยเฉพาะ ปัจจุบันการบำบัดด้วยยีนสามารถทำได้ในเยอรมนีเท่านั้น ขณะนี้มีผู้ป่วยเพียงไม่กี่รายที่ได้รับการรักษาด้วยยานี้
อาหารและไลฟ์สไตล์
ผู้ที่เป็นโรคธาลัสซีเมียสามารถส่งผลดีต่อการเกิดโรคได้โดยการรับประทานอาหารที่เหมาะสมและดำเนินชีวิตอย่างเหมาะสม โปรดทราบสิ่งต่อไปนี้:
- กินอาหารที่สมดุล.
- หลีกเลี่ยงอาหารที่มีธาตุเหล็กสูงมาก (เช่น ตับ)
- รับแคลเซียมจากอาหารเพียงพอ (เช่น นม โยเกิร์ต ชีส ใบผักโขม บร็อคโคลี่)
- อย่าดื่มแอลกอฮอล์
- ออกกำลังกายเป็นประจำ (ออกกำลังกายประมาณ 3-4 ชั่วโมง เช่น เดินเร็ว วิ่งเหยาะๆ หรือปั่นจักรยานต่อสัปดาห์)
โรคธาลัสซีเมียมีอาการอย่างไร?
อาการจะรุนแรงเพียงใดนั้นขึ้นอยู่กับว่าฮีโมโกลบินมีการเปลี่ยนแปลงอย่างผิดปกติมากน้อยเพียงใด ผู้ที่เป็นธาลัสซีเมียที่ไม่รุนแรงมักมีอาการเพียงเล็กน้อยหรือไม่มีเลย
อย่างไรก็ตาม หากไม่ได้รับการรักษา ทารกที่เป็นโรคธาลัสซีเมียแบบรุนแรงจะมีปัญหาสุขภาพตั้งแต่อายุสี่หรือห้าเดือน
โรคโลหิตจาง
ผู้ที่เป็นโรคธาลัสซีเมียที่สำคัญหรือคนกลางมักแสดงอาการของโรคโลหิตจาง:
- มีสีซีดหรือมีผิวสีเหลือง
- คุณเหนื่อยตลอดเวลา
- คุณรู้สึกเวียนหัว
- คุณมีอาการปวดหัว
- คุณมีการเต้นของหัวใจอย่างรวดเร็ว
- พวกเขาหายใจไม่ออกอย่างรวดเร็วระหว่างการออกกำลังกาย
- พวกเขามักจะมีตับและม้ามโต
- การพัฒนาของพวกเขามักจะถูกรบกวน
เมื่อมองแวบแรก ธาลัสซีเมียมีลักษณะคล้ายกับภาวะโลหิตจางจากการขาดธาตุเหล็ก อย่างไรก็ตาม ระดับธาตุเหล็กจะต่ำในกรณีของการขาดธาตุเหล็ก ในขณะที่เป็นปกติในโรคธาลัสซีเมีย
ภายหลังหรือหากการรักษาไม่เพียงพอ ผู้ที่ได้รับผลกระทบมักพบผลข้างเคียงที่รุนแรง ซึ่งรวมถึงตัวอย่างเช่น:
- ความผิดปกติของกระดูก
- ติดเชื้อบ่อย
- ปัญหาหัวใจ
- โรคเบาหวาน
- โรคกระดูกพรุน
การเปลี่ยนแปลงของกระดูก
ผู้ที่เป็นโรคธาลัสซีเมียที่สำคัญหรือตัวกลางมักแสดงการเติบโตของกระดูกผิดปกติ:
- กระดูกกะโหลกศีรษะขยายใหญ่ขึ้น
- หน้าผาก กรามบน และกระดูกโหนกแก้มโป่งพอง
- ซี่โครงและกระดูกสันหลังงอ
- คุณมีความเสี่ยงที่จะกระดูกหักเพิ่มขึ้น
- คุณมีอาการปวดกระดูก
- คุณเป็นโรคกระดูกพรุน
ธาตุเหล็กส่วนเกิน
ผู้ที่เป็นโรคธาลัสซีเมียแบบรุนแรงมักเก็บกักธาตุเหล็กในร่างกายมากเกินไป (ธาตุเหล็กเกิน) ซึ่งจะไม่สลายไปเองอีกต่อไป สิ่งนี้สามารถทำลายอวัยวะ (hemochromatosis ทุติยภูมิ) ธาตุเหล็กที่เพิ่มขึ้นนั้นมาจากเลือดส่วนเกินที่ผู้ได้รับผลกระทบได้รับเป็นประจำโดยการฉีด
นอกจากนี้ ร่างกายยังถือว่าโรคโลหิตจางเป็นภาวะขาดธาตุเหล็กและพยายามชดเชยด้วยการบริโภคธาตุเหล็กจากอาหารมากขึ้น ร่างกายยังผลิตเซลล์เม็ดเลือดมากเกินไป ตับและม้ามทำงานเต็มที่เพื่อสลายอีกครั้ง
ความเข้มข้นของธาตุเหล็กที่สูงเกินไปทำให้เกิดอาการอื่น ๆ ในผู้ที่ได้รับผลกระทบเช่น:
- ภาวะหัวใจล้มเหลว (หัวใจล้มเหลว)
- หัวใจเต้นผิดจังหวะ
- ความผิดปกติของตับ
- โรคเบาหวาน (เบาหวาน)
- ขนาดสั้น
- วัยแรกรุ่นล่าช้า
- ต่อมไทรอยด์ทำงานน้อย (พร่อง)
- ความผิดปกติของการเผาผลาญวิตามินดี
เมื่อไปพบแพทย์
เนื่องจากรูปแบบที่รุนแรงของธาลัสซีเมียมักปรากฏขึ้นในช่วงสองสามเดือนแรกของชีวิต เป็นสิ่งสำคัญที่พ่อแม่ควรพบกุมารแพทย์โดยเร็วที่สุดที่สัญญาณแรกเริ่ม (เช่น ซีด)
คุณจะป้องกันได้อย่างไร?
แพทย์แนะนำให้ผู้ที่รู้ว่าตนเองมียีนสำหรับธาลัสซีเมียให้ไปที่ศูนย์ให้คำปรึกษาทางพันธุกรรมก่อนกำหนดการตั้งครรภ์หรือหากต้องการมีบุตร
ผู้เชี่ยวชาญที่ผ่านการฝึกอบรมใช้การตรวจเลือดทางพันธุกรรมเพื่อกำหนดความเสี่ยงทางพันธุกรรมที่เป็นไปได้ที่เกี่ยวข้องกับการตั้งครรภ์ในคู่รัก
ผู้ที่มีประวัติครอบครัวเป็นธาลัสซีเมียควรขอคำแนะนำก่อนตั้งครรภ์
เพื่อป้องกันการติดเชื้อที่คุกคามชีวิต แพทย์ยังแนะนำให้ฉีดวัคซีนเด็กที่เป็นโรคธาลัสซีเมียและเด็กที่มีสุขภาพแข็งแรงตามปฏิทินการฉีดวัคซีนปัจจุบัน ในบางกรณี จำเป็นต้องรักษาเด็กที่เป็นโรคธาลัสซีเมียด้วยยาปฏิชีวนะบางชนิดเป็นประจำ สิ่งเหล่านี้ช่วยป้องกันการติดเชื้อแบคทีเรียร้ายแรง ซึ่งผู้ที่เป็นโรคธาลัสซีเมียจะอ่อนแอเป็นพิเศษ
การวัดอุณหภูมิร่างกายเป็นประจำจะช่วยตรวจจับการติดเชื้อได้เร็วที่สุด และรักษาได้โดยเร็วที่สุด หากมีไข้เกิน 38.5 องศาเซลเซียส ควรไปพบแพทย์ทันที เป็นไปได้ว่าการติดเชื้อทำให้เกิดสิ่งนี้
นอกจากนี้ยังเป็นสิ่งสำคัญโดยเฉพาะอย่างยิ่งที่ผู้ได้รับผลกระทบจะได้รับการตรวจสอบอย่างสม่ำเสมอจากแพทย์ นี่เป็นวิธีเดียวที่จะติดตามอาการของโรคอย่างระมัดระวังและระบุและรักษาภาวะแทรกซ้อนในระยะเริ่มแรก
ธาลัสซีเมียรักษาได้หรือไม่?
ปัจจุบันการรักษาธาลัสซีเมียแบบสมบูรณ์สามารถทำได้ด้วยการปลูกถ่ายเซลล์ต้นกำเนิดหรือการบำบัดด้วยยีน อายุขัยและคุณภาพชีวิตของผู้ที่ได้รับผลกระทบเพิ่มขึ้นอย่างต่อเนื่องจากยาตัวใหม่ การรักษาที่ดีขึ้น และการศึกษาที่ดีขึ้น
การพยากรณ์โรคธาลัสซีเมียเป็นอย่างไร?
ผู้ที่เป็นธาลัสซีเมียที่ไม่รุนแรง (อัลฟ่าเล็กน้อยหรือเบต้าธาลัสซีเมีย) มีอายุขัยเฉลี่ย ตามกฎแล้ว ผู้ได้รับผลกระทบสามารถอยู่ได้โดยปราศจากการด้อยค่า
ด้วยรูปแบบที่รุนแรงกว่านั้น ก็มักจะเป็นไปได้ที่จะบรรลุอายุขัยที่เกือบจะปกติด้วยการรักษาที่ถูกต้องและตั้งแต่เนิ่นๆ (เช่น การถ่ายเลือดเป็นประจำ) อย่างไรก็ตาม การบำบัดตลอดชีวิต ต่อเนื่อง และเข้มข้นเป็นสิ่งจำเป็น
บางครั้งการปลูกถ่ายสเต็มเซลล์สามารถรักษาโรคได้ และในบางกรณีที่พบไม่บ่อยอาจต้องใช้ยีนบำบัด
แท็ก: สัมภาษณ์ กายวิภาคศาสตร์ เด็กทารก