กล้ามเนื้อลีบกระดูกสันหลัง
และ Florian Tiefenböck คุณหมอ อัปเดตเมื่อMaximilian Reindl ศึกษาวิชาเคมีและชีวเคมีที่ LMU ในมิวนิก และเป็นสมาชิกของทีมบรรณาธิการของ ตั้งแต่เดือนธันวาคม 2020 เขาจะทำความคุ้นเคยกับหัวข้อนโยบายทางการแพทย์ วิทยาศาสตร์ และสุขภาพสำหรับคุณ เพื่อให้เข้าใจและเข้าใจได้
โพสต์เพิ่มเติมโดย Maximilian ReindlFlorian Tiefenböck ศึกษาการแพทย์ของมนุษย์ที่ LMU มิวนิก เขาเข้าร่วม ในฐานะนักเรียนในเดือนมีนาคม 2014 และได้สนับสนุนทีมบรรณาธิการด้วยบทความทางการแพทย์ตั้งแต่นั้นเป็นต้นมา หลังจากได้รับใบอนุญาตทางการแพทย์และการปฏิบัติงานด้านอายุรศาสตร์ที่โรงพยาบาลมหาวิทยาลัยเอาก์สบูร์ก เขาได้เป็นสมาชิกถาวรของทีม ตั้งแต่เดือนธันวาคม 2019 และเหนือสิ่งอื่นใด ยังรับประกันคุณภาพทางการแพทย์ของเครื่องมือ
กระทู้เพิ่มเติมโดย Florian Tiefenböck เนื้อหา ทั้งหมดได้รับการตรวจสอบโดยนักข่าวทางการแพทย์
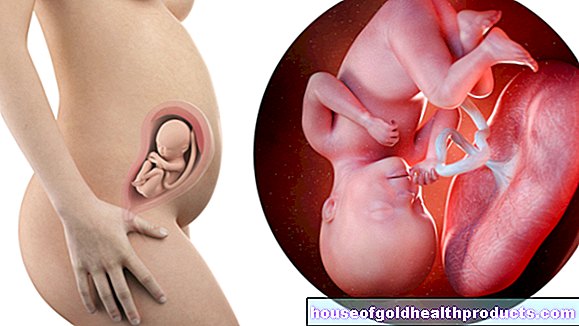
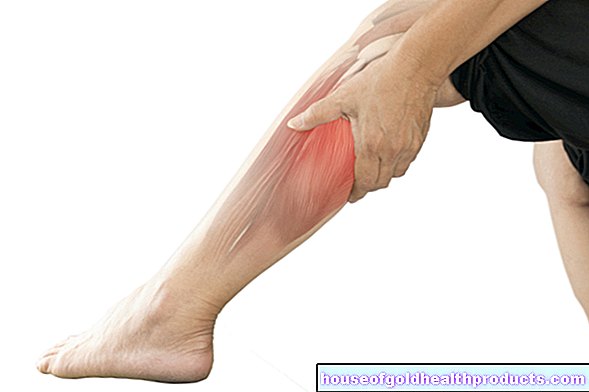
กล้ามเนื้อลีบกระดูกสันหลังหรือ SMA เรียกสั้น ๆ ว่าเป็นโรคที่หายากซึ่งเซลล์ประสาทบางชนิดในไขสันหลังอักเสบตาย แรงกระตุ้นและแรงกระตุ้นจากสมองไปไม่ถึงจุดหมายอีกต่อไป นั่นคือ กล้ามเนื้อ ทำให้สูญเสียกล้ามเนื้อและเป็นอัมพาต SMA มีหลายรูปแบบ ที่รุนแรงที่สุดเริ่มต้นในวัยเด็ก การรักษาแบบใหม่ช่วยให้มีสุขภาพที่ดีขึ้นได้อย่างยั่งยืน อ่านเพิ่มเติมเกี่ยวกับการฝ่อของกล้ามเนื้อกระดูกสันหลังได้ที่นี่
รหัส ICD สำหรับโรคนี้: รหัส ICD เป็นรหัสที่เป็นที่ยอมรับในระดับสากลสำหรับการวินิจฉัยทางการแพทย์ สามารถพบได้เช่นในจดหมายของแพทย์หรือในใบรับรองความสามารถในการทำงาน G12

ภาพรวมโดยย่อ
- กล้ามเนื้อลีบกระดูกสันหลังคืออะไร? กลุ่มโรคกล้ามเนื้ออ่อนแรง ขึ้นอยู่กับการตายของเซลล์ประสาทบางชนิดในไขสันหลังที่ควบคุมกล้ามเนื้อ (เซลล์ประสาทสั่งการ) ดังนั้น SMA จึงเป็นหนึ่งในโรคของเซลล์ประสาทสั่งการ
- มีแบบฟอร์มอะไรบ้าง? กล้ามเนื้อลีบของกระดูกสันหลังโดยกรรมพันธุ์ส่วนใหญ่เป็น SMA ที่มีข้อบกพร่องทางพันธุกรรมบางอย่างในโครโมโซม 5 (5q-associated SMA) แพทย์แยกแยะระหว่างสี่รูปแบบที่แตกต่างกัน: SMA ประเภท 1 - ประเภท 4 นอกจากนี้ยังมีรูปแบบประปรายซึ่งไม่แน่นอนทางพันธุกรรม
- ความถี่: โรคหายาก; SMA ที่สืบทอดมามีผลกระทบต่อทารกแรกเกิดหนึ่งคนในปี 7000
- อาการ: กล้ามเนื้อกระตุก, กล้ามเนื้ออ่อนแรง, การสูญเสียกล้ามเนื้อ, อัมพาต การไล่ระดับสีจะแตกต่างกันไปตามรูปร่าง SMA
- สาเหตุ: กล้ามเนื้อลีบที่กระดูกสันหลังโดยกรรมพันธุ์ 1-4 เป็นผลมาจากความบกพร่องทางพันธุกรรมในโครโมโซม 5 ซึ่งแม่นยำกว่าในยีน SMN1 ส่งผลให้ร่างกายขาดโปรตีนพิเศษ คือ โปรตีน SMN การขาดดุลนี้ทำลายเซลล์ประสาทสั่งการในไขสันหลัง
- การวินิจฉัย: การตรวจทางพันธุกรรมสำหรับการสร้างพันธุกรรม SMN ที่เปลี่ยนแปลงไป การตรวจร่างกาย อิเล็กโตรโนกราฟี อิเล็กโตรไมโอกราฟี การตรวจเลือด (เช่น CK)
- การรักษา: การบำบัดทดแทนยีนหรือการบริหารยาของโมดูเลเตอร์ประกบกันเป็นไปได้ กายภาพบำบัดควบคู่ การบำบัดด้วยการพูด การบำบัดความเจ็บปวด และจิตบำบัด หากจำเป็นให้ทำการผ่าตัดกระดูกสันหลัง แผนการรักษาขึ้นอยู่กับประเภทของ SMA
- การพยากรณ์โรค: ในกรณีของ SMA ใกล้เคียงที่ถ่ายทอดทางพันธุกรรม ตัวเลือกการรักษาแบบใหม่มีผลเชิงสาเหตุและอาจส่งผลในเชิงบวกต่อการเกิดโรค การเริ่มต้นการรักษาตั้งแต่เนิ่นๆเป็นสิ่งสำคัญ การรักษายังไม่พร้อมสำหรับผู้ป่วยทุกราย หากไม่ได้รับการรักษา เด็กที่เป็นโรค SMA ประเภท 1 มักจะเสียชีวิตภายในสองปีแรก อายุขัยในประเภทที่ 3 และประเภทที่ 4 แทบจะไม่ลดลงเลย
กล้ามเนื้อลีบกระดูกสันหลังคืออะไร?
ในการฝ่อของกล้ามเนื้อกระดูกสันหลัง (SMA) เซลล์ประสาทบางเซลล์ในไขสันหลังจะตาย พวกเขามักจะควบคุมกล้ามเนื้อซึ่งเป็นสาเหตุที่ผู้เชี่ยวชาญเรียกเซลล์ประสาทเหล่านี้ว่าเป็นเซลล์ประสาทสั่งการ ดังนั้น SMA จึงเป็นโรคที่เรียกว่าโรคเซลล์ประสาทสั่งการ
ในกรณีของกล้ามเนื้อลีบกระดูกสันหลัง เซลล์ประสาทสั่งการส่วนล่าง (ที่สอง) ซึ่งเชื่อมต่อโดยตรงกับกล้ามเนื้อด้วยส่วนต่อของพวกมันจะได้รับผลกระทบ อันเป็นผลมาจากความเสียหาย สัญญาณประสาทจะไปถึงกล้ามเนื้อน้อยลงหรือไม่มีเลย กล้ามเนื้ออ่อนแรงลงเรื่อยๆ (กล้ามเนื้อลีบ / กล้ามเนื้อลีบ)
แพทย์แยกแยะความแตกต่างระหว่างรูปแบบต่างๆ ของกล้ามเนื้อลีบกระดูกสันหลัง กลุ่มที่ใหญ่ที่สุดคือ SMA ทางพันธุกรรมซึ่งส่งผลกระทบต่อกล้ามเนื้อใกล้กับลำตัว (ส่วนใกล้เคียง) ขึ้นอยู่กับข้อบกพร่องทางพันธุกรรมที่เฉพาะเจาะจง ทารกแรกเกิดประมาณ 1 ใน 7,000 คนจะพัฒนามัน
กล้ามเนื้อลีบกระดูกสันหลังเป็นโรคที่หายากโดยรวม อย่างไรก็ตาม โรคนี้เป็นโรคที่ถ่ายทอดทางพันธุกรรมโดย autosomal recessive ที่พบได้บ่อยเป็นอันดับ 2 และยังเป็นสาเหตุการเสียชีวิตที่พบบ่อยที่สุดในทารกหรือเด็กวัยหัดเดินเนื่องจากความบกพร่องทางพันธุกรรม
กล้ามเนื้อลีบของกระดูกสันหลังมีกี่ประเภท?
แพทย์แยกแยะรูปแบบทางพันธุกรรม (กรรมพันธุ์) ของ SMA จากรูปแบบประปราย การจำแนกประเภทอื่นของกล้ามเนื้อลีบเกี่ยวกับกระดูกสันหลังส่วนใหญ่เกี่ยวข้องกับกลุ่มกล้ามเนื้อที่ได้รับผลกระทบก่อน มี
- Proximal SMA: ด้วยประมาณ 90 เปอร์เซ็นต์ พวกเขาสร้างกลุ่ม SMA ที่ใหญ่ที่สุด อาการเริ่มต้นที่กล้ามเนื้อใกล้ลำตัว กล่าวคือ ใกล้เคียงกัน
- Non-proximal SMA: ที่นี่ กลุ่มกล้ามเนื้อที่อยู่ห่างไกลออกไป เช่น มือและเท้า จะได้รับผลกระทบก่อน (SMA ส่วนปลาย) ในระยะต่อไป SMA เหล่านี้ยังสามารถแพร่กระจายไปยังกล้ามเนื้อบริเวณกลางลำตัวได้อีกด้วย
- รูปแบบพิเศษ (เช่น โรคกล้ามเนื้อลีบ Spinobulbar ชนิด Kennedy)
กล้ามเนื้อลีบของกระดูกสันหลังส่วนต้น
กล้ามเนื้อลีบของกระดูกสันหลังส่วนปลายโดยกรรมพันธุ์มักเป็นโรคที่เกิดจากความบกพร่องทางพันธุกรรมเฉพาะ (SMA ที่เกี่ยวข้องกับ 5q, ข้อบกพร่องในโครโมโซม 5) สิ่งเหล่านี้จะถูกแบ่งออกเป็นสี่รูปแบบที่แตกต่างกัน การจำแนกประเภทขึ้นอยู่กับเวลาที่อาการแรกปรากฏขึ้นและตามหลักสูตรของโรค
Spinal Muscular Atrophy Type 1: นี่เป็นรูปแบบ SMA ที่พบได้บ่อยและร้ายแรงที่สุด เรียกอีกอย่างว่า "โรค Werdnig-Hoffmann" หรือ "SMA เด็กอ่อนเฉียบพลัน" โรคนี้มักเริ่มในวัยเด็กตอนต้น ความอ่อนแอของกล้ามเนื้อส่งผลต่อร่างกายทั้งหมด - แพทย์ยังพูดถึง "กลุ่มอาการของทารกที่อ่อนแอ" (จากภาษาอังกฤษ floppy = อ่อนแอ, ทารก = ทารก, เด็ก) เด็กที่ไม่ได้รับการรักษาที่มี SMA ประเภท 1 ส่วนใหญ่เสียชีวิตก่อนอายุสองขวบ
กล้ามเนื้อลีบกระดูกสันหลังประเภทที่ 2: รูปแบบของ SMA นี้เรียกอีกอย่างว่า "กล้ามเนื้อลีบกระดูกสันหลังระดับกลาง" หรือ "SMA ในวัยแรกเกิดเรื้อรัง" อาการแรกมักเกิดขึ้นก่อนอายุ 18 เดือน ผู้ที่ได้รับผลกระทบอาจมีอายุขัยลดลงอย่างมีนัยสำคัญในบางครั้ง
กล้ามเนื้อลีบกระดูกสันหลังประเภทที่ 3: มันยังเป็นที่รู้จักกันในนาม "โรคกล้ามเนื้อลีบกระดูกสันหลังเด็กและเยาวชน" หรือ "โรค Kugelberg-Welander" SMA นี้มักจะเริ่มหลังจากอายุ 18 เดือนและก่อนวัยผู้ใหญ่ตอนต้น กล้ามเนื้ออ่อนแรงน้อยกว่าในประเภท 1 หรือ 2 ผู้ที่ได้รับผลกระทบมีอายุขัยลดลงเพียงเล็กน้อยเท่านั้น
กล้ามเนื้อลีบกระดูกสันหลังประเภทที่ 4: คล้ายกับ SMA ประเภท 3 แต่ปรากฏเฉพาะในวัยผู้ใหญ่ (โดยปกติ> 30 ปี) อย่างไรก็ตาม ความอ่อนแอของกล้ามเนื้อมีความเด่นชัดน้อยกว่าและดำเนินไปช้ากว่า SMA ประเภท 3
การเปลี่ยนระหว่างเวอร์ชันต่างๆ เป็นแบบไหล ในบางกรณี การทำเช่นนี้จะทำให้การแบ่งเขตที่ชัดเจนทำได้ยาก ความบกพร่องทางพันธุกรรมบางอย่างยังมีบทบาทสำคัญในความรุนแรงของโรคที่เป็นปัญหา
กล้ามเนื้อลีบกระดูกสันหลังอื่นๆ
นอกจากรูปแบบใกล้เคียงเหล่านี้แล้ว ยังมีรูปแบบอื่นๆ ของการฝ่อของกล้ามเนื้อกระดูกสันหลัง เหล่านี้รวมถึง ตัวอย่างเช่น หายากกว่า ลีบของกล้ามเนื้อกระดูกสันหลังส่วนปลายที่ถ่ายทอดทางพันธุกรรม กับพวกเขา อาการมักจะเริ่มต้นในกลุ่มกล้ามเนื้อที่อยู่ไกลจากร่างกาย
ไม่รับประกันมรดกในกรณีของ SMA ประปราย นอกจากนี้ยังไม่สามารถระบุการสะสมของครอบครัวได้ ในวรรณคดี ได้แก่
- ประเภทฮิรายามะ (SMA ปลายเด็กและเยาวชน โรคอายุประมาณ 15 ปี ส่งผลต่อกล้ามเนื้อแขน มักจะหยุดนิ่งแม้จะไม่มีการรักษาและสามารถปรับปรุงได้)
- ประเภท Vulpian-Bernhard (รวมถึงกลุ่มอาการ "แขนตีบ" โดยเริ่มมีอาการที่ผ้าคาดไหล่ โดยปกติเมื่ออายุ 40 ปีขึ้นไป)
- ประเภท Duchenne-Aran (เริ่มแรกกล้ามเนื้อมือแผ่กระจายไปยังลำตัวโดยปกติหลังจากอายุ 30 ปี)
- ประเภท Peroneal (กลุ่มอาการ "flail-leg" ครั้งแรกที่กล้ามเนื้อขาส่วนล่าง)
- Progressive bulbar palsy (ความผิดปกติของการพูดและการกลืนมีผลต่อประมาณ 20 เปอร์เซ็นต์ของผู้ป่วยที่มีเส้นโลหิตตีบด้านข้าง amyotrophic)
รูปแบบ SMA ประปราย (กลุ่มอาการ "แขนขา - / - ขา", อัมพาต bulbar แบบก้าวหน้า) นับรวมอยู่ในกลุ่มของเส้นโลหิตตีบด้านข้าง amyotrophic (ALS) ในแวดวงผู้เชี่ยวชาญ บทความนี้เกี่ยวข้องกับการฝ่อของกล้ามเนื้อกระดูกสันหลังส่วนต้นทางพันธุกรรม
กล้ามเนื้อลีบ Spinobulbar
กล้ามเนื้อลีบ Spinobulbar หรือ bulbospinal (ประเภท Kennedy, Kennedy syndrome) เป็นโรคที่สืบทอดมา มักเริ่มตั้งแต่วัยหนุ่มสาวจนถึงวัยกลางคน แบบฟอร์ม SMA พิเศษนี้สืบทอดมาจากลักษณะถอย X-linked และมีผลกับผู้ชายเท่านั้น (เนื่องจากผู้ชายมีโครโมโซม X เพียงอันเดียว ในผู้หญิงจะมีโครโมโซม X ตัวที่สองที่มีสุขภาพดีและจะชดเชยข้อบกพร่อง)
กล้ามเนื้ออ่อนแรงในกล้ามเนื้อใกล้กับร่างกายที่ขาและแขนหรือไหล่ตลอดจนกล้ามเนื้อลิ้นและลำคอเป็นเรื่องปกติ ส่งผลให้ผู้ที่ได้รับผลกระทบมีปัญหาในการพูดและการกลืน เป็นต้น พวกเขายังบ่นถึงอาการสั่น ปวดกล้ามเนื้อ และกระตุก ผู้ชายที่ได้รับผลกระทบมักจะมีลูกอัณฑะแคระแกรนและเป็นหมัน นอกจากนี้ ต่อมน้ำนมขยายใหญ่ขึ้น (gynecomastia)
กล้ามเนื้อลีบ Spinobulbar มักจะช้า อายุขัยแทบจะไม่ถูกจำกัด
คุณรู้จักกล้ามเนื้อลีบกระดูกสันหลังได้อย่างไร?
โดยทั่วไปของกล้ามเนื้อลีบกระดูกสันหลังคือกล้ามเนื้ออ่อนแรงแบบก้าวหน้าจนถึงอัมพาต (อัมพฤกษ์) และการกระตุกของกล้ามเนื้อ อันเป็นผลมาจากความเสียหายของเส้นประสาท กล้ามเนื้อไม่ได้รับแรงกระตุ้นไฟฟ้าอีกต่อไป ซึ่งทำให้พวกมันหดตัวเมื่อเวลาผ่านไป (กล้ามเนื้อลีบ) สัญญาณและข้อร้องเรียนที่แน่นอนขึ้นอยู่กับแบบฟอร์มที่เกี่ยวข้อง ส่วนต่อไปนี้จะกล่าวถึงอาการของ SMA ใกล้เคียงทางพันธุกรรม
อาการของกล้ามเนื้อลีบกระดูกสันหลังในวัยแรกเกิดประเภท 1
ด้วย SMA type 1 อาการจะปรากฏขึ้นในช่วงหกเดือนแรกของชีวิต กล้ามเนื้ออ่อนแรงทั่วไปเกิดขึ้น - นั่นคือสิ่งที่ส่งผลกระทบต่อทั้งร่างกาย นอกจากนี้ความตึงเครียดระหว่างกล้ามเนื้อจะลดลง แพทย์พูดถึงภาวะ hypotonia
ในทารกแรกเกิด ความอ่อนแอของกล้ามเนื้อนี้เริ่มปรากฏในท่าขาทั่วไป ซึ่งชวนให้นึกถึงกบที่กำลังนอนอยู่ (ท่าขากบ) ขางอเข่าทำมุมออกด้านนอกและเท้าทำมุมเข้าด้านใน แม้แต่การยกหรือจับศีรษะด้วยตัวเองก็มักจะทำไม่ได้
ในวัยสูงอายุ เด็กที่เป็นโรค SMA ประเภท 1 ไม่สามารถนั่งหรือเดินเองได้ เด็กหลายคนยังพูดไม่ได้ เนื่องจากอาจส่งผลต่อกล้ามเนื้อลิ้นได้
อีกลักษณะหนึ่งของการลีบของกล้ามเนื้อกระดูกสันหลังแบบที่ 1 คือรูปร่างของร่างกายส่วนบน: กล้ามเนื้อของหน้าอกและหลังไม่พัฒนาอย่างเหมาะสม สิ่งนี้ทำให้ร่างกายส่วนบนมีรูปร่างเหมือนระฆัง (หน้าอกระฆัง) เนื่องจากการพัฒนาของกล้ามเนื้อบริเวณหน้าอกและหลังที่ต่ำ ผู้ที่ได้รับผลกระทบจึงมีท่าที่โค้ง
มักจะมีความโค้งของกระดูกสันหลังเพิ่มขึ้น (scoliosis) ท่าทางที่ค่อมไปข้างหน้าและหลังค่อมทำให้เกิดปัญหาการหายใจเพิ่มเติม หายใจเร็วและตื้นมาก (หายใจเร็ว) เป็นลักษณะเฉพาะ
อาการของกล้ามเนื้อลีบกระดูกสันหลังระดับกลางประเภท2
กล้ามเนื้อลีบกระดูกสันหลังชนิดที่ 2 มักทำให้เกิดอาการระหว่างอายุเจ็ดถึง 18 เดือนเท่านั้น เด็กที่ได้รับผลกระทบสามารถนั่งได้เอง แต่มักจะไม่เรียนรู้ที่จะยืนหรือเดิน กล้ามเนื้ออ่อนแรงดำเนินไปช้ากว่าแบบที่ 1
สำหรับ SMA ชนิดที่ 2 อาการคล้ายกับอาการของทารกในวัยแรกเกิดที่รุนแรง เช่น กระดูกสันหลังผิดรูป เกิดขึ้นเมื่อเวลาผ่านไป ข้อต่อแข็งขึ้นเนื่องจากกล้ามเนื้อและเส้นเอ็นสั้นลง (การหดตัว) อาการอื่นๆ ได้แก่ อาการสั่นที่มือและการกระตุกของกล้ามเนื้อในลิ้น
อาการของกล้ามเนื้อลีบกระดูกสันหลังเด็กและเยาวชนประเภท 3
กล้ามเนื้อลีบกระดูกสันหลังประเภทที่ 3 มักปรากฏขึ้นหลังจากอายุ 18 เดือนและก่อนอายุ 18 ปี เด็กที่ได้รับผลกระทบสามารถนั่ง ยืน และเดินได้อย่างอิสระ อย่างไรก็ตาม กล้ามเนื้ออ่อนแรง โดยเฉพาะกล้ามเนื้ออุ้งเชิงกรานและขา ทำให้เกิดการเดินเตาะแตะ
ในช่วงหลายปีที่ผ่านมาประสิทธิภาพลดลง: ในตอนแรกผู้ที่ได้รับผลกระทบพบว่ากิจกรรมกีฬาหรือปีนบันไดยาก แต่สุดท้ายก็ยากที่จะพกถุงช้อปปิ้งเป็นต้น หลังจากผ่านไปหลายปี กล้ามเนื้อลีบที่กระดูกสันหลังแบบที่ 3 ทำให้การวิ่งและการออกแรงอื่นๆ ทำได้ยากหรือเป็นไปไม่ได้ แม้แต่ในผู้สูงอายุ
อย่างไรก็ตาม โดยรวมแล้ว อาการต่างๆ มีความเด่นชัดน้อยกว่าในโรคอื่นๆ อีก 2 รูปแบบ คือ ชนิดที่ 1 และชนิดที่ 2 สำหรับผู้ป่วยจำนวนมากที่ได้รับผลกระทบ คุณภาพชีวิตแทบจะไม่ลดลงในระยะเวลานาน
อาการของกล้ามเนื้อลีบในผู้ใหญ่ประเภท 4
การสูญเสียกล้ามเนื้อแบบก้าวหน้ารูปแบบที่เกิดขึ้นได้ยากนี้เริ่มต้นขึ้นในวัยผู้ใหญ่ ซึ่งมักเกิดขึ้นในช่วงทศวรรษที่สามของชีวิต กล้ามเนื้อขาและสะโพกได้รับผลกระทบในขั้นต้น เมื่อโรคดำเนินไป กล้ามเนื้ออ่อนแรงก็กระจายไปที่ไหล่และแขน
อาการของภาพทางคลินิกคล้ายกับของ SMA เด็กและเยาวชนประเภท 3 อย่างไรก็ตาม กล้ามเนื้ออ่อนแรงแบบก้าวหน้านั้นช้ากว่า SMA ประเภท 3
อะไรทำให้กล้ามเนื้อลีบของกระดูกสันหลัง?
ในการฝ่อของกล้ามเนื้อกระดูกสันหลัง เซลล์ประสาทสั่งการที่สองในไขสันหลังจะพินาศ เหล่านี้เป็นเซลล์ประสาทที่ควบคุมกล้ามเนื้อด้วยอวัยวะ อันเป็นผลมาจากความเสียหายต่อเซลล์ประสาทสั่งการที่มีความเชี่ยวชาญสูงเหล่านี้ สัญญาณไฟฟ้าที่ส่งถึงกล้ามเนื้อน้อยลงกว่าในคนที่มีสุขภาพดี หากเซลล์กล้ามเนื้อถูกใช้น้อยลงและถูกกระตุ้นน้อยลง ร่างกายก็จะสลายตัวเมื่อเวลาผ่านไป
ข้อบกพร่องทางพันธุกรรม
ในกรณีส่วนใหญ่ กล้ามเนื้อลีบกระดูกสันหลังเป็นโรคทางพันธุกรรม (กรรมพันธุ์ SMA) สาเหตุของรูปแบบ SMA ใกล้เคียงโดยทั่วไปคือข้อมูลที่ไม่ถูกต้องในองค์ประกอบทางพันธุกรรมของผู้ป่วย ยีนที่เรียกว่า SMN1 บนโครโมโซม 5 ไม่ทำงาน
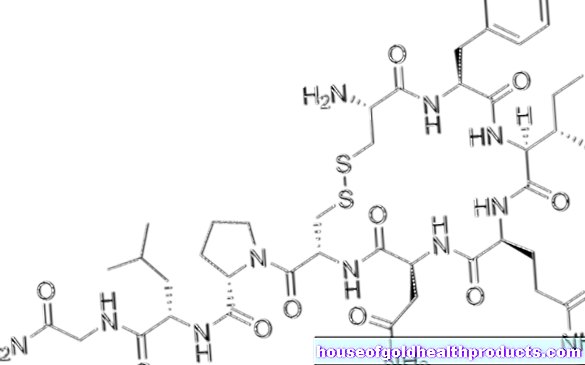
ยีน SMN1 นำข้อมูล - เช่น พิมพ์เขียว - สำหรับโมเลกุลโปรตีนที่สำคัญที่เรียกว่า SMN SMN ย่อมาจาก "Survival (of) Motor Neuron" หากไม่มีโมเลกุลโปรตีน SMN เซลล์ประสาทสั่งการก็จะพินาศตามกาลเวลา
เป็นความจริงที่ว่ายังมียีน SMN2 ที่เกี่ยวข้องในร่างกาย ซึ่งโดยหลักการแล้วสามารถ "ชดเชย" สำหรับข้อมูลทางพันธุกรรม SMN1 ที่ไม่ทำงานได้ แต่สิ่งนี้มักจะเกิดขึ้นเพียงเล็กน้อยเท่านั้น ซึ่งหมายความว่าการสูญเสียการทำงานของยีน SMN1 (หากไม่ได้รับการรักษา) มักจะไม่สามารถชดเชยได้อย่างสมบูรณ์โดยสำเนายีน SMN2 ที่ไม่บุบสลาย
autosomal recessive และ autosomal dominant inheritance
ข้อมูลทางพันธุกรรมของบุคคลนั้นซ้ำกัน ด้วยเหตุนี้ ทุกคนจึงมียีน SMN1 สองชุด สำเนาหนึ่งมาจากพ่อและอีกชุดหนึ่งมาจากแม่ กล้ามเนื้อลีบของกระดูกสันหลังส่วนต้นในวัยเด็กมักได้รับการถ่ายทอดเป็นลักษณะถอยแบบ autosomal
ซึ่งหมายความว่ายีนทั้งสองแบบ (อัลลีล) จากพ่อแม่ต้องมีข้อบกพร่องในการฝ่อของกล้ามเนื้อกระดูกสันหลังเพื่อพัฒนาในลูกหลาน ในกรณีของการถ่ายทอดทางพันธุกรรมแบบด้อย พ่อแม่จะไม่ได้รับผลกระทบเพราะนอกจากยีนที่ไม่ทำงานแล้ว พวกเขายังมียีน SMN1 ที่มีสุขภาพดีซึ่งชดเชยข้อบกพร่องอีกด้วย
ทุกๆ คนที่ 45 เป็นเจ้าของระบบนี้สำหรับ SMA คู่สมรสที่ทั้งคู่เป็นพาหะมีความเสี่ยง 25% ที่จะมีลูกที่เป็นโรคนี้
ในบางกรณีในวัยรุ่น การฝ่อของกล้ามเนื้อกระดูกสันหลังในวัยผู้ใหญ่โดยเฉพาะอย่างยิ่งยังเป็นไปตามการถ่ายทอดทางพันธุกรรมที่เด่นชัดของ autosomal ในกรณีของการถ่ายทอดทางพันธุกรรมที่ครอบงำ ยีนที่บกพร่องได้ยืนยันตัวเองแล้ว - และผู้ที่ได้รับผลกระทบจะป่วย อย่างไรก็ตาม นี่ไม่ใช่ข้อบกพร่องทางพันธุกรรมที่กล่าวถึงข้างต้นในโครโมโซม 5 SMA ที่เกี่ยวข้องกับ 5q เหล่านี้มักสืบทอดมาในลักษณะด้อยแบบ autosomal
การสืบทอดกับรูปแบบ SMA อื่น ๆ
นอกจากนี้ยังสามารถสืบทอดกล้ามเนื้อลีบของกระดูกสันหลังส่วนปลายได้ รูปแบบ spinobulbar พิเศษ (ชนิด Kennedy) ได้รับการถ่ายทอดทางโครโมโซมเพศซึ่งเป็นโครโมโซม X (ซึ่งส่งผลต่อยีนที่มีพิมพ์เขียวสำหรับไซต์เทียบท่าสำหรับฮอร์โมนเพศชาย) ในกรณีของรูปแบบประปรายอย่างไรก็ตามไม่รับประกันมรดก ไม่ค่อยมีใครรู้จักที่นี่ว่าทำไมเซลล์ประสาทสั่งการตัวที่สองถึงพินาศ
การตรวจและวินิจฉัย
การวินิจฉัยโรคกล้ามเนื้อลีบกระดูกสันหลังมักทำโดยกุมารแพทย์ กุมารแพทย์ที่เชี่ยวชาญในโรคเส้นประสาท (neuropediatricians) และผู้เชี่ยวชาญด้านโรคของระบบประสาท (นักประสาทวิทยา) การทดสอบต่างๆ จำเป็นสำหรับการชี้แจงที่แม่นยำยิ่งขึ้น ในกรณีของ SMA การทดสอบทางพันธุกรรมและการตรวจเส้นประสาทและกล้ามเนื้อมีความสำคัญเป็นพิเศษ
รวบรวมประวัติการรักษา (anamnesis)
ในทุกอาการป่วย แพทย์จะถามถึงอาการที่เกิดขึ้นก่อนและว่าจนถึงปัจจุบันเป็นอย่างไร ในทารกและเด็กเล็ก ผู้ปกครองรายงานการเปลี่ยนแปลงและความผิดปกติในพฤติกรรมของลูก โดยเฉพาะโรคทางพันธุกรรม แพทย์ยังให้ความสำคัญกับประวัติทางการแพทย์ของครอบครัวด้วย
การตรวจร่างกาย
โดยทั่วไป แพทย์จะกำหนดความผิดปกติในการพัฒนายนต์โดยการตรวจร่างกายเด็ก ตัวอย่างเช่น การทดสอบว่าเด็กๆ สามารถตั้งศีรษะ นั่งหรือขยับแขนหรือขาได้อย่างอิสระหรือไม่ (ขึ้นอยู่กับอายุ)
การทดสอบการออกกำลังกายเสริมจะดำเนินการในเด็กโตและผู้ใหญ่ที่สงสัยว่ามีกล้ามเนื้อลีบกระดูกสันหลัง แพทย์จะตรวจสอบว่าผู้ที่เกี่ยวข้องสามารถรวบรวมกำลังได้มากเพียงใดและสามารถรับได้นานแค่ไหน เขายังตรวจสอบความอดทน
นอกจากนี้ แพทย์จะทดสอบปฏิกิริยาตอบสนอง ซึ่งโดยปกติแล้วจะอ่อนแรงหรือดับลง โดยเฉพาะอย่างยิ่งในกรณีที่กล้ามเนื้อลีบของกระดูกสันหลังที่เด่นชัด เมื่อต้องการทำเช่นนี้ เขาใช้ค้อนเคาะเส้นเอ็นต่างๆ เช่น ที่ส้นเท้าหรือใต้เข่า แล้วตรวจดูปฏิกิริยา
การศึกษาทางพันธุกรรม
วิธีการตรวจหาที่น่าเชื่อถือที่สุดสำหรับการฝ่อของกล้ามเนื้อกระดูกสันหลัง (กรรมพันธุ์) คือ การวิเคราะห์ทางพันธุกรรม แพทย์จะมองหาหลักฐานของยีน SMN1 ที่เปลี่ยนแปลง (กลายพันธุ์) และจำนวนสำเนา SMN2 ที่มีอยู่
กฎทั่วไปคือการวินิจฉัยและรักษา (กรรมพันธุ์) SMA ให้เร็วที่สุด ขึ้นอยู่กับรูปแบบและการรักษาที่มีอยู่ การพัฒนามอเตอร์สามารถได้รับอิทธิพลในทางบวกก่อนที่เซลล์ประสาทสั่งการของไขสันหลังจะเสียหายอย่างถาวร
การสอบสวนเพิ่มเติมที่SMA
หากสงสัยว่าเป็น SMA แพทย์มักจะวัดความเร็วการนำของเส้นประสาท (electroneurography) และกิจกรรมของกล้ามเนื้อ (electromyography) หากจำเป็น พวกเขายังตรวจกล้ามเนื้อโดยใช้อัลตราซาวนด์ (myosonography) หรือการถ่ายภาพด้วยคลื่นสนามแม่เหล็ก (MRI)
นอกจากนี้แพทย์จะทำการตรวจเลือด หากมีกล้ามเนื้อลีบของกระดูกสันหลัง พารามิเตอร์บางอย่างสามารถเปลี่ยนแปลงได้ ตัวอย่างเช่น ระดับของไคเนสของครีเอทีน (CK ซึ่งเป็นเอนไซม์ของกล้ามเนื้อทั่วไป) จะเพิ่มขึ้น
การรักษากล้ามเนื้อลีบกระดูกสันหลัง
การรักษากล้ามเนื้อลีบของกระดูกสันหลังนั้นซับซ้อน เป็นเวลานาน การรักษาเชิงสาเหตุไม่สามารถทำได้สำหรับ SMA ทุกรูปแบบ อย่างไรก็ตาม ความก้าวหน้าในการวิจัยทางการแพทย์ช่วยให้แพทย์มีทางเลือกในการรักษาใหม่ๆ เพื่อช่วยเหลือผู้ที่ได้รับผลกระทบจาก proximal SMA (ข้อบกพร่องของยีน SMN บนโครโมโซม 5) โดยพื้นฐานแล้ว
นอกจากนี้ แพทย์ยังให้ความสำคัญกับการบรรเทาอาการและให้การสนับสนุนที่ดีที่สุดแก่ผู้ที่ได้รับผลกระทบ (เช่น กายภาพบำบัด การบำบัดทางเดินหายใจ จิตบำบัด การผ่าตัด)
การรักษาพยาบาล
แนวทางการรักษาแบบใหม่สำหรับผู้ป่วยที่ SMA มีพื้นฐานมาจากข้อบกพร่องของยีน SMN ที่รู้จักนั้นแทรกแซงโดยตรงในสารพันธุกรรมเองหรือในการประมวลผลข้อมูลทางพันธุกรรมที่ปลายน้ำ
จุดมุ่งหมายคือเพื่อให้ร่างกายของผู้ป่วยสามารถผลิตโปรตีน SMN ในปริมาณที่เพียงพอได้อย่างอิสระ ซึ่งเป็นสิ่งสำคัญสำหรับเซลล์ประสาทสั่งการ
มีตัวเลือกการรักษาต่อไปนี้สำหรับการฝ่อของกล้ามเนื้อกระดูกสันหลัง:
- โมดูเลเตอร์ Splicing (Nusinersen, Risdiplam): ยาเหล่านี้แทรกแซงโดยตรงในการประมวลผลโมเลกุล RNA ของผู้ส่งสารโดยตรง พวกเขาเสริมสร้างกระบวนการเหล่านั้นที่ส่งโปรตีน SMN ในปริมาณที่สูงขึ้นจากยีน SMN2 ที่ไม่บุบสลาย
- การบำบัดทดแทนยีน (Onasemnogene Abeparvovec): การบำบัดนี้แทรกแซงโดยตรงในจีโนมของมนุษย์ สำเนาที่ผิดพลาดของยีน SMN1 จะถูกแทนที่ในเซลล์ที่ได้รับผลกระทบด้วยโครงสร้างยีนที่ทำหน้าที่จัดหาจากภายนอก
โมดูเลเตอร์ประกบ
ในกรณีของยีน SMN1 บกพร่อง ร่างกายสามารถผลิตโปรตีน SMN จากยีน SMN2 ที่เกี่ยวข้องได้ ยีนทดแทน SMN2 "กระโดดเข้ามา" แต่นั่นไม่เพียงพอ เหตุผล: โปรตีนใน SMN2 มักจะสั้นเกินไปและแตกตัวอย่างรวดเร็ว
นี่เป็นเพราะการประมวลผลของ SMN2 messenger RNA ที่สอดคล้องกัน (SMN2 mRNA) มันส่งข้อมูลการก่อสร้างจากจีโนม (DNA) ไปยังไซต์การผลิตโปรตีน (ไรโบโซม)
ในการทำเช่นนี้ ยีน SMN2 ในจีโนมจะถูกอ่านก่อน มีการสร้าง RNA ผู้ส่งสาร SMN2 เบื้องต้น เหนือสิ่งอื่นใด มันต้องดำเนินการเพิ่มเติมผ่านการประกบที่เรียกว่า จากนั้น RNA ของผู้ส่งสารที่โตเต็มที่ก็เกิดขึ้นเท่านั้น คอมเพล็กซ์เซลล์พิเศษคือไรโบโซม จากนั้นอ่าน RNA ของผู้ส่งสารที่เจริญเต็มที่แล้วจึงผลิตโปรตีน SMN2 และนี่คือสิ่งที่สั้นลงและไม่เสถียร ถูกรื้อถอนอย่างรวดเร็ว และไม่สามารถเข้าควบคุมฟังก์ชันของ SMN1 ได้
เพื่อเปลี่ยนแปลงสิ่งนี้ ส่วนผสมออกฤทธิ์ Nusinersen และ Risdiplam มีอิทธิพลต่อการประมวลผลต่อไปของ RNA ผู้ส่งสารเบื้องต้น ด้วยเหตุนี้ โมดูเลเตอร์ splicing modulators เหล่านี้จึงเพิ่มปริมาณโปรตีน SMN ที่ใช้งานได้ในที่สุด และทำให้มั่นใจได้ว่ามีอุปทานเพียงพอ
Nusinersen
ยา Nusinersen เป็นสิ่งที่เรียกว่า "antisense oligonucleotide" (ASO) ได้รับการอนุมัติจาก European Medicines Agency ในปีพ. ศ. 2560 ASO ถูกผลิตขึ้นโดยเทียมและดัดแปลงโมเลกุล RNA เป็นพิเศษ พวกเขาผูกมัดอย่างเฉพาะเจาะจงและแม่นยำกับ RNA ของผู้ส่งสาร SMN2 สิ่งนี้จะป้องกันไม่ให้ถูกประมวลผลอย่างไม่ถูกต้องในเซลล์ของมนุษย์
โดยเฉพาะ: Nusinersen ป้องกันไม่ให้ข้อมูลสำคัญ (exon 7) ถูกตัดออกจาก SMN2 messenger RNA อย่างผิดพลาด ตำแหน่งของ exon 7 ทำให้ร่างกายสร้างโปรตีน SMN ที่ทำงานได้ดีขึ้นในภายหลัง
Nusinersen ได้รับผ่านสิ่งที่เรียกว่าการเจาะเอว ซึ่งหมายความว่ายาถูกฉีดเข้าไปในคลองกระดูกสันหลังด้วยเข็มฉีดยา การบำบัดนี้จะทำซ้ำเป็นระยะ ๆ เป็นเวลาสองสามเดือน ในปีแรกของการรักษา ผู้ที่ได้รับผลกระทบจะได้รับ 6 ครั้ง จากนั้น 3 ครั้งต่อปี
ผู้ป่วยมักจะทนต่อยาได้ดี Nusinersen นำไปสู่หลักสูตรโรคที่เอื้ออำนวยมากขึ้น จากการศึกษาพบว่าผู้ป่วยจำนวนมากสามารถเคลื่อนไหวได้: ในหลายกรณี สามารถนั่งได้อย่างอิสระและหมุนร่างกายได้อย่างอิสระ ผลข้างเคียงและภาวะแทรกซ้อนขึ้นอยู่กับการเจาะเอว (เช่น ปวดศีรษะ การติดเชื้อที่เยื่อหุ้มสมอง)
ริสดิพลัม
คณะกรรมาธิการยุโรปอนุมัติ Risdiplam เป็นยาตัวที่สามกับ SMA ที่เกี่ยวข้องกับ 5q (ประเภท 1-3 หรือสำเนายีน SMN2) หนึ่งถึงสี่ชุด) ในเดือนมีนาคม 2564 Risdiplam นำมาทุกวันเป็นผงที่ละลายน้ำได้ ปริมาณที่แน่นอนคำนวณจากอายุและน้ำหนักตัว
Risdiplam ไม่ใช่ "antisense oligonucleotide" ซึ่งแตกต่างจาก Nusinersen แต่เป็นโมเลกุลขนาดเล็ก โมเลกุลนี้จับกับ RNA ของผู้ส่งสารสำหรับโปรตีน SMN2 และทำให้เสถียรในลักษณะนี้ เป็นผลให้มีการสร้างโปรตีน SMN ที่ใช้งานได้มากขึ้น
ผลข้างเคียงที่พบบ่อยของ Risdiplam ได้แก่ ความรู้สึกไม่สบายในทางเดินอาหาร ผื่น มีไข้ และการติดเชื้อทางเดินปัสสาวะ
การบำบัดทดแทนยีน
อีกวิธีหนึ่งในการรักษากล้ามเนื้อลีบของกระดูกสันหลังส่วนต้นนั้นขึ้นอยู่กับสิ่งที่เรียกว่าการบำบัดทดแทนยีน ยีน SMN1 ที่บกพร่อง ซึ่งเป็นจุดเริ่มต้นของโปรเกรสซี SMA ถูก "แทนที่" ด้วยสำเนาฟังก์ชันใหม่ของยีน
สารออกฤทธิ์ Onasemnogene Abeparvovec (AVXS-101) ซึ่งทำงานบนหลักการนี้ ได้รับอนุญาตทางการตลาดแบบมีเงื่อนไขสำหรับการรักษาเด็กวัยหัดเดินและเด็กจาก European Medicines Agency (EMA) ในเดือนพฤษภาคม 2020
ยานี้สามารถใช้สำหรับ SMA ชนิดที่ 1 ตามข้อมูล EMA ในรูปแบบอื่นทั้งหมดของโรค SMA ลักษณะทางพันธุกรรม (จำนวนสำเนา SMN2) ตัดสินใจว่าการบำบัดด้วยการทดแทนยีนเป็นทางเลือกหนึ่งหรือไม่
ด้วย Onasemnogene Abeparvovec สำเนาการทำงานของยีน SMN1 ของมนุษย์จะถูกนำเข้าสู่เซลล์ที่ได้รับผลกระทบของไขสันหลังและก้านสมอง สิ่งนี้ทำได้โดยไวรัสบางชนิดที่ทำหน้าที่เป็น "เรือข้ามฟาก" สำหรับสารพันธุกรรมใหม่ - ที่เรียกว่าเวกเตอร์ไวรัสที่เกี่ยวข้องกับ adeno (AAV vectors)
การสร้างยีนเวกเตอร์จะได้รับครั้งเดียวโดยการฉีดผ่านหลอดเลือดดำเข้าสู่กระแสเลือดและจากที่นั่นจะกระจายไปทั่วร่างกาย เนื่องจากตัวกั้นเลือดและสมองยังพัฒนาไม่เต็มที่ในเด็กเล็ก พาหะเหล่านี้จึงสามารถเข้าไปในเนื้อเยื่อไขสันหลังได้
โดยเฉพาะอย่างยิ่งการจับเวกเตอร์เหล่านี้กับโครงสร้างพื้นผิวพิเศษของเซลล์ประสาทสั่งการ พวกมันจึงเลือกสารพันธุกรรมขึ้นมาเพื่อผลิตโปรตีน SMN อย่างอิสระ
การรักษาสามารถปรับปรุงการทำงานของมอเตอร์และนำไปสู่ความสำเร็จในการพัฒนาอย่างยั่งยืน (เช่น การนั่ง การคลาน และการเดินโดยไม่ได้รับการสนับสนุน)ในระหว่างการรักษา ค่าตับบางครั้งอาจเพิ่มขึ้นอย่างมีนัยสำคัญ แต่จำนวนเกล็ดเลือดสามารถลดลงได้ ไข้และอาเจียนเป็นเรื่องปกติ เพื่อลดผลข้างเคียง ผู้ป่วยจะได้รับคอร์ติโคสเตียรอยด์ ("คอร์ติโซน") เป็นเวลาสองสามสัปดาห์
โดยทั่วไปแล้วการพัฒนามอเตอร์ให้เหมาะสมกับวัยจะเกิดขึ้นได้ก็ต่อเมื่อการบำบัดด้วยยีนเริ่มต้นโดยไม่ได้แสดงอาการ การรักษาเกิดขึ้นในศูนย์บำบัดโรคประสาทและกล้ามเนื้อเฉพาะทาง
กายภาพบำบัด
กายภาพบำบัดยังคงเป็นเสาหลักที่สำคัญในการรักษา SMA ไม่ใช่ทุกรูปแบบของ SMA ที่สามารถรักษาได้ด้วยวิธีการรักษาแบบใหม่ การบำบัดด้วยการออกกำลังกายเป็นประจำได้รับการออกแบบมาเพื่อรักษาความสามารถทางกายภาพและชะลอการสลายตัวของกล้ามเนื้อ
นักกายภาพบำบัดเคลื่อนไหวส่วนต่าง ๆ ของร่างกายที่เป็นอัมพาตไปแล้วอย่างอดทน ลำดับการเคลื่อนไหวที่เคลื่อนไหวได้รับการฝึกฝนเพื่อรองรับการเคลื่อนไหวและความแข็งแรงของกล้ามเนื้อ การนวดหรือการประคบร้อนและเย็นก็สามารถช่วยได้เช่นกัน สิ่งเหล่านี้ยังช่วยผ่อนคลายและชะลอความเสื่อมลงอีกในบางกรณี
การบำบัดด้วยการพูด
ในบางกรณี SMA ส่งผลต่อการพูดและการกลืนกล้ามเนื้อ จากนั้นการออกกำลังกายบำบัดด้วยการพูดจะช่วยได้ ส่งเสริมให้เด็กเรียนรู้ที่จะพูด แม้แต่ในผู้ป่วยสูงอายุ ก็มักจะทำให้การพูดเสื่อมช้าลงได้ นักบำบัดด้วยการพูดยังฝึกการกลืนที่ถูกต้องอีกด้วย
นักกายภาพบำบัดและนักบำบัดการพูดสนับสนุนผู้ที่ได้รับผลกระทบจากการบำบัดด้วยการหายใจแบบกำหนดเป้าหมาย
ยาแก้ปวด
การบำบัดด้วยความเจ็บปวดมีบทบาทสำคัญ โดยเฉพาะอย่างยิ่งในระยะขั้นสูงของโรค แพทย์ใช้ยาคลายความเจ็บปวดเพื่อลดความทุกข์ทรมานของผู้ได้รับผลกระทบ
การผ่าตัด
เนื่องจากกล้ามเนื้อลีบของกระดูกสันหลังอาจนำไปสู่ความโค้งของกระดูกสันหลังอย่างรุนแรง (scoliosis) แพทย์จึงพิจารณาการผ่าตัดในบางครั้ง การทำเช่นนี้จะทำให้กระดูกสันหลังแข็งขึ้นโดยเฉพาะ
สิ่งนี้ทำให้ผู้ที่ได้รับผลกระทบมีความมั่นคงของลำตัว (บางส่วน) มากขึ้น ซึ่งไม่เพียงแต่ช่วยให้มีท่าทางตั้งตรงมากขึ้น แต่ยังช่วยปกป้องกระดูกและข้อต่ออีกด้วย การผ่าตัดกระดูกสันหลังยังสามารถช่วยป้องกันปัญหาการหายใจแบบก้าวหน้าได้
การดูแลจิตบำบัด
โรคทางประสาทและกล้ามเนื้อ เช่น กล้ามเนื้อลีบกระดูกสันหลังแสดงถึงความเครียดทางจิตใจ ผู้ป่วยและญาติจะดำเนินการวินิจฉัยในรายบุคคลและกลุ่มที่นำโดยจิตบำบัดและพัฒนากลยุทธ์เพื่อจัดการกับโรคได้ดีขึ้น
กลุ่มช่วยเหลือตนเองและตัวแทนผู้ป่วยยังให้การสนับสนุนที่สำคัญอีกด้วย พวกเขาแจ้ง ให้คำแนะนำ และสนับสนุนผู้ที่ได้รับผลกระทบและญาติของพวกเขาเพื่อรับมือกับความท้าทายของโรค SMA
โอกาสในการฟื้นตัวจากกล้ามเนื้อลีบกระดูกสันหลัง
หากมีกล้ามเนื้อลีบที่กระดูกสันหลัง การพยากรณ์โรคจะขึ้นอยู่กับรูปร่างเป็นหลัก ยิ่งมีอาการมากเท่าไรก็ยิ่งดีขึ้นเท่านั้น นอกจากนี้ แพทย์ก่อนหน้านี้วินิจฉัยว่ากล้ามเนื้อลีบของกระดูกสันหลัง ยิ่งพวกเขาสามารถเริ่มมาตรการการรักษาที่เหมาะสมได้เร็วเท่านั้น แม้กระทั่งก่อนที่เซลล์ประสาทสั่งการจะถูกทำลายอย่างถาวร
ตัวเลือกการรักษาใหม่ผ่านโมดูเลเตอร์ splicing modulators และการบำบัดทดแทนยีนมีศักยภาพที่ดีในการรักษา SMA ใกล้เคียง โดยเฉพาะอย่างยิ่งเมื่อเริ่มการรักษา (มาก) แต่เนิ่นๆ อย่างไรก็ตาม ข้อมูลสำหรับการพยากรณ์โรคในระยะยาวที่เชื่อถือได้ยังคงรอดำเนินการอยู่ เฉพาะการศึกษาเพิ่มเติมและการสังเกตความปลอดภัยของยาอย่างใกล้ชิดเท่านั้นที่สามารถให้ความแน่นอนเพิ่มเติมในอีก (เดือนและ) ปีถัดไป ด้วยยาที่ใหม่กว่า อย่างน้อยก็สามารถควบคุมโรคในระยะยาวหรือแม้แต่การรักษาได้
โดยทั่วไป SMA ประเภท 1 เป็นโรคร้ายแรง เด็กที่พัฒนา SMA ประเภท 1 มี (ไม่ได้รับการรักษา) อายุขัยที่ จำกัด มาก ความอ่อนแอของกล้ามเนื้อที่เพิ่มขึ้นอย่างรวดเร็วทั่วร่างกายก็ส่งผลต่อการหายใจเช่นกัน ผลที่ตามมาคือปอดบวมเฉียบพลันและแม้แต่การหายใจล้มเหลว เด็กที่ได้รับผลกระทบเสียชีวิตภายในสองสามปีแรกของชีวิต
การพยากรณ์โรคดีขึ้นเล็กน้อยสำหรับ SMA type 2 อายุขัยเฉลี่ยแตกต่างกันไปขึ้นอยู่กับความรุนแรงของโรค: บางคนเสียชีวิตในวัยเด็ก แต่ส่วนใหญ่ถึงวัยหนุ่มสาว ไม่ช้าก็เร็ว - ถ้าต้องการ - การหายใจจะต้องได้รับการสนับสนุนในรูปแบบที่รุนแรงมากขึ้น ผู้ที่ได้รับผลกระทบสามารถเคลื่อนที่ได้โดยใช้รถเข็น
ด้วย SMA ประเภท 3 การพยากรณ์โรคจะดีขึ้นอย่างมาก - โดยเฉพาะอย่างยิ่งถ้าอาการแรกเกิดขึ้นช้า ประสิทธิภาพการทำงานค่อยๆเสื่อมลงในช่วงหลายปีที่ผ่านมา ในวัยชราอาจจำเป็นต้องใช้รถเข็นหรือการดูแลอย่างถาวร อายุขัยแทบจะไม่ถูกจำกัดโดยกล้ามเนื้อลีบกระดูกสันหลังชนิดที่ 3
กล้ามเนื้อลีบของกระดูกสันหลังในผู้ใหญ่ (ประเภทที่ 4) นั้นช้ากว่าประเภทที่ 3 ด้วยซ้ำ ผู้คนมักมีอายุขัยเฉลี่ย
แท็ก: tcm เด็กทารก เด็กวัยหัดเดิน