พราเดอร์-วิลลี่ ซินโดรม
Clemens Gödel เป็นฟรีแลนซ์ให้กับทีมแพทย์ของ
ข้อมูลเพิ่มเติมเกี่ยวกับผู้เชี่ยวชาญของ เนื้อหา ทั้งหมดได้รับการตรวจสอบโดยนักข่าวทางการแพทย์กลุ่มอาการ Prader-Willi (PWS) เป็นผลมาจากข้อบกพร่องที่มีมา แต่กำเนิดในสารพันธุกรรม ทารกที่ได้รับผลกระทบนั้นสั้น ด้อยพัฒนาทางจิตใจ และมีกล้ามเนื้อ ในวัยเด็กพวกเขาพัฒนาความหิวที่ไม่รู้จักพอซึ่งนำไปสู่โรคอ้วนที่เด่นชัด โรคที่เกิดขึ้นจะต้องได้รับการรักษาโดยการรักษาจากแพทย์เฉพาะทางต่างๆ อ่านเพิ่มเติมเกี่ยวกับอาการ การวินิจฉัย และการรักษาโรค Prader-Willi ได้ที่นี่!
รหัส ICD สำหรับโรคนี้: รหัส ICD เป็นรหัสที่เป็นที่ยอมรับในระดับสากลสำหรับการวินิจฉัยทางการแพทย์ สามารถพบได้เช่นในจดหมายของแพทย์หรือในใบรับรองความสามารถในการทำงาน Q87
กลุ่มอาการ Prader-Willi: คำอธิบาย
กลุ่มอาการ Prader-Willi-Syndrome (ผิดด้วย Willi-Prader-Syndrome) ได้รับการอธิบายเป็นครั้งแรกในปี 1956 โดยกุมารแพทย์ Andrea Prader, Alexis Labhart และ Heinrich Willi ทารกแรกเกิดประมาณ 1 ใน 20,000 คนเป็นโรคพราเดอร์-วิลลี่ สาเหตุคือความผิดปกติที่กระตุ้นทางพันธุกรรมของไฮโปทาลามัส ซึ่งเป็นศูนย์กลางการสับเปลี่ยนที่สำคัญในสมอง อาการของโรค Prader-Willi อาจแตกต่างกันมากและซับซ้อน
กลุ่มอาการพราเดอร์-วิลลี่: อาการ
ทารกในครรภ์ที่ได้รับผลกระทบจะสังเกตเห็นได้แม้กระทั่งก่อนเกิด พวกมันเคลื่อนไหวเพียงเล็กน้อยในครรภ์อย่างเห็นได้ชัด อัตราการเต้นของหัวใจต่ำกว่าปกติ ในช่วงคลอด ทารกในครรภ์ที่มีอาการ Prader-Willi มักจะอยู่ในตำแหน่งที่ผิดปกติในร่างกายของมารดา ทารกต้องการการสนับสนุนอย่างมากระหว่างและหลังคลอด
ทันทีหลังคลอด ทารกแรกเกิดที่ได้รับผลกระทบมีความโดดเด่นเนื่องจากการใช้ชีวิตอยู่ประจำ กล้ามเนื้ออ่อนแรง และน้ำหนักแรกเกิดต่ำ เสียงกรีดร้องทั่วไปหลังคลอดอาจหายไปหรืออ่อนแอเท่านั้น เนื่องจากความอ่อนแอที่เด่นชัดและความผิดปกติในการดูดและกลืนทำให้ทารกดื่มได้ยาก
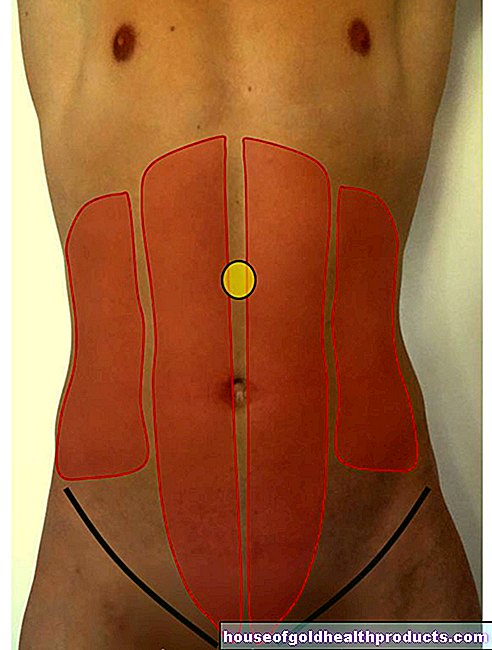
ทารกที่มีอาการ Prader-Willi มักมีลักษณะภายนอกบางอย่าง ใบหน้าแคบมีลักษณะเป็นตารูปอัลมอนด์และปากเล็กที่มีริมฝีปากบนบาง กะโหลกศีรษะมักจะยาว (dolidochocephaly) และมือและเท้ามีขนาดเล็ก กระดูกสันหลังสามารถโค้งงอเป็นรูปตัว S (scoliosis) สารกระดูกในร่างกายแสดงความเสียหายและข้อบกพร่อง (โรคกระดูกพรุน / ภาวะกระดูกพรุน) ในภาพเอ็กซ์เรย์ เม็ดสีของผิวหนัง เส้นผม และเรตินาอาจลดลง การรบกวนทางสายตาและตำแหน่งเหล่ (ตาเหล่) ของดวงตาก็เป็นไปได้เช่นกัน ถุงอัณฑะมีขนาดเล็กและมักว่างเปล่า (ลูกอัณฑะที่ไม่ได้รับ) โดยรวมแล้วพัฒนาการของเด็กป่วยล่าช้า
ในตอนท้ายของปีแรกของชีวิตความอ่อนแอของกล้ามเนื้อจะดีขึ้นบ้าง อย่างไรก็ตาม อย่างน้อยก็มีจุดอ่อนที่ไม่รุนแรงอยู่เสมอ กิจกรรมปกติอาจทำให้ผู้ป่วยที่เป็นโรค Prader-Willi syndrome เหนื่อยและต้องใช้กำลังมาก ถึงกระนั้น เด็ก ๆ ก็สนุกกับกิจกรรมในวัยเด็กทั่วไปพอๆ กัน การเจริญเติบโตจะลดลงอย่างมากในวัยเตาะแตะ
การรับประทานอาหารที่ไม่ถูกยับยั้ง
ในขณะที่โรคดำเนินไป เด็กที่ได้รับผลกระทบจะกินมากขึ้น (hyperphagia) - โดยไม่รู้สึกอิ่ม อาหารถูกกักตุนไว้และแม้ในกรณีที่ร้ายแรงก็ถูกขโมย เป็นเรื่องยากมากสำหรับเด็กที่จะควบคุมนิสัยการกินของพวกเขา ดังนั้นพวกเขาจึงพัฒนาโรคอ้วน (โรคอ้วน) อย่างชัดเจน พวกเขามีน้ำหนักเพิ่มขึ้นอย่างรวดเร็วโดยเฉพาะอย่างยิ่งเนื่องจากมีความคลาดเคลื่อนระหว่างการบริโภคแคลอรี่สูงและการใช้พลังงานต่ำ
การมีน้ำหนักเกินทำให้เกิดโรครองตามมาคือ หัวใจและปอดต้องทนทุกข์ทรมานจากความเครียดจากโรคอ้วน หนึ่งในสี่ของผู้ที่ได้รับผลกระทบทั้งหมดเป็นโรคเบาหวานเมื่ออายุ 20 ปี ความผิดปกติของการนอนหลับ โรคหลอดเลือดดำ (thrombophlebitis) และการกักเก็บน้ำก็เป็นหนึ่งในผลกระทบที่อาจเกิดขึ้น ในระหว่างที่เป็นโรคนี้ อาการนอนไม่หลับอาจเกิดขึ้นได้ นอกจากการหยุดหายใจซ้ำแล้วซ้ำเล่า อาจเกิดการรบกวนจังหวะกลางวัน-กลางคืน หรือการนอนหลับสนิทได้ เด็กที่ได้รับผลกระทบพบว่าการเรียนรู้ยาก
พัฒนาการของวัยแรกรุ่นถูกรบกวน
การเจริญเติบโตโดยทั่วไปในช่วงวัยแรกรุ่นมีขนาดเล็ก เด็กที่ได้รับผลกระทบมักไม่สูงเกิน 140 ถึง 160 เซนติเมตร แม้ว่าวัยแรกรุ่นสามารถเริ่มก่อนวัยอันควร (adrenarche ก่อนวัยอันควร) ในหลายกรณีการพัฒนาของวัยแรกรุ่นไม่สิ้นสุด ผู้ที่ได้รับผลกระทบมักจะเป็นหมัน ในเด็กผู้ชาย องคชาตและโดยเฉพาะอัณฑะยังเล็กอยู่ ไม่มีการสร้างเม็ดสีและริ้วรอยบนถุงอัณฑะ เสียงแตกอาจไม่เกิดขึ้น ในเด็กผู้หญิง ริมฝีปากและอวัยวะเพศหญิงยังคงด้อยพัฒนา การมีประจำเดือนครั้งแรกไม่เกิดขึ้นเลย ทั้งก่อนเวลาอันควรหรือช่วงปลายๆ บางครั้งอาจอยู่ในช่วงอายุระหว่าง 30 ถึง 40 ปีเท่านั้น
การพัฒนาจิตใจและจิตใจ
ทั้งการพัฒนาทางจิตและจิตประสาทถูกรบกวนในกลุ่มอาการ Prader-Willi เหตุการณ์สำคัญในการพัฒนาเด็กมักเกิดขึ้นช้ากว่าเพื่อนที่มีสุขภาพดี การพัฒนาทางภาษาศาสตร์และการเคลื่อนไหวบางครั้งใช้เวลานานเป็นสองเท่าของเพื่อนที่มีสุขภาพดี
ความฉลาดทางสติปัญญาเฉลี่ย (IQ) อยู่ระหว่าง 60 ถึง 70 และต่ำกว่าเกณฑ์ปกติมาก ความอ่อนแอทางร่างกายอาจทำให้การพูดยากขึ้น การพัฒนาคำพูดล่าช้าและความเข้าใจคำพูดบกพร่อง ขึ้นอยู่กับ IQ ต่ำ ประมาณ 40 เปอร์เซ็นต์ของผู้ป่วยใกล้จะพิการทางสติปัญญาแล้ว ความผิดปกติของการเรียนรู้เช่นปัญหาเลขคณิตก็ปรากฏขึ้นโดยไม่ขึ้นกับไอคิว ผลงานของโรงเรียนส่วนใหญ่ต่ำกว่าค่าเฉลี่ย
สังเกตได้ทั้งพัฒนาการทางอารมณ์และพฤติกรรม บางครั้งคนที่ได้รับผลกระทบมักถูกมองว่าดื้อรั้นและอารมณ์อ่อนไหว ความผิดปกติทางจิตเวชสามารถเกิดขึ้นได้ในวัยเด็กตอนต้น: มีการอธิบายรูปแบบของพฤติกรรมที่เรียกว่าฝ่ายตรงข้ามตลอดจนพฤติกรรมที่เข้มงวดและเป็นเจ้าของ กระบวนการประจำอาจเป็นเรื่องยาก บางครั้งกระบวนการต้องทำซ้ำอย่างบังคับ ลักษณะออทิสติกเกิดขึ้นในประมาณ 25 เปอร์เซ็นต์ของผู้ป่วย โรคสมาธิสั้น (ADD) ก็เป็นเรื่องปกติเช่นกัน
อาการมักจะเพิ่มขึ้นตามอายุและน้ำหนักเกิน อย่างไรก็ตาม ในผู้สูงอายุ อาการ Prader-Willi syndrome สามารถถดถอยได้ง่าย ประมาณร้อยละสิบต้องทนทุกข์ทรมานจากโรคจิต นอกจากนี้ โรคลมบ้าหมูและรูปแบบของ "การเสพติดการนอน" (narcolepsy) มีความเกี่ยวข้องกับกลุ่มอาการพราเดอร์-วิลลี่
Prader-Willi syndrome: สาเหตุและปัจจัยเสี่ยง
สาเหตุของโรค Prader-Willi เชื่อกันว่าเป็นความผิดปกติของสมองส่วนกลางที่เรียกว่าไฮโปทาลามัส เหนือสิ่งอื่นใด สิ่งนี้ส่งผลให้เกิดการขาดฮอร์โมนการเจริญเติบโตที่สำคัญ ความผิดปกติเกิดขึ้นประมาณสามในสี่ของกรณีโดยขาดส่วนของยีนในโครโมโซม 15 (15q11-q13) ในกลุ่มอาการ Prader-Willi ข้อบกพร่องในสำเนาโครโมโซมของบิดาดูเหมือนจะมีความสำคัญเป็นพิเศษ (70 ถึง 75 เปอร์เซ็นต์ ) เพื่อให้มียีนเพียงสำเนาเดียว ความเป็นไปได้อีกประการหนึ่งคือยีนทั้งสองชุดของโครโมโซมคู่นั้นมาจากแม่เท่านั้น (ความผิดปกติแบบผู้ปกครองเดียว 25 ถึง 30 เปอร์เซ็นต์) สิ่งที่เรียกว่า “รอยตำหนิ” นั้นพบได้น้อยกว่า (ร้อยละหนึ่ง) คำว่า "การประทับ" อธิบายถึงความจริงที่ว่ายีนถูกอ่านขึ้นอยู่กับที่มาของยีน (ในด้านมารดาหรือบิดา)
ในกรณีส่วนใหญ่ โรคนี้ไม่สามารถถ่ายทอดทางพันธุกรรมได้ มักเกิดขึ้นระหว่างการพัฒนาเซลล์สืบพันธุ์หรือหลังการปฏิสนธิเท่านั้น ในทางกลับกัน เป็นไปได้ว่าการกลายพันธุ์ของยีนที่มีอยู่ (ส่วนใหญ่เรียกว่าการโยกย้ายที่สมดุล) ก็ทำให้เกิดกลุ่มอาการ Prader-Willi ในกรณีเหล่านี้ความเสี่ยงของการสืบทอดจะเพิ่มขึ้น
Prader-Willi Syndrome: การสอบสวนและการวินิจฉัย
ทารกแรกเกิดทุกคนที่มีความอ่อนแออย่างต่อเนื่องและไม่ได้อธิบายควรได้รับการทดสอบสำหรับ PWS แม้แต่แพทย์ทารกแรกเกิด (neonatologist) หรือกุมารแพทย์ (กุมารแพทย์) จะสงสัยกลุ่มอาการ Prader-Willi หลังคลอดเนื่องจากพฤติกรรมของเด็ก ไม่มีการทดสอบเชิงทำนายที่เรียกว่าดำเนินการโดยไม่มีหลักฐานว่ามีอาการนี้ อย่างไรก็ตาม การวินิจฉัยกลุ่มอาการ Prader-Willi มีความเป็นไปได้ค่อนข้างมากก่อนคลอด
การตรวจร่างกายและเทคนิค
ในกรณีส่วนใหญ่ การตรวจร่างกายทำให้เกิดความสงสัยในระดับสูงเกี่ยวกับกลุ่มอาการ Prader-Willi (Holms Criteria 1993, 2001) ลักษณะสำคัญของกลุ่มอาการ Prader-Willi คือความอ่อนแอที่เด่นชัดซึ่งเห็นได้ชัดโดยเฉพาะอย่างยิ่งเมื่อดื่ม ลักษณะที่ปรากฏยังให้เบาะแส โดยปกติการตอบสนองที่ตรวจพบได้นั้นเด่นชัดเพียงเล็กน้อยเท่านั้น
การขาดฮอร์โมนการเจริญเติบโตในเลือดที่วัดได้ก็มีประโยชน์ในการวินิจฉัยเช่นกัน ฮอร์โมนเพศ (เอสโตรเจน, เทสโทสเตอโรน, FSH, LH) ส่วนใหญ่ลดลงในผู้ที่ได้รับผลกระทบเช่นกัน สิ่งนี้มาพร้อมกับความล้าหลังของอวัยวะสืบพันธุ์ การทำงานของเยื่อหุ้มสมองต่อมหมวกไตถูกรบกวนในหลายกรณี การก่อตัวของฮอร์โมนเพศ (แอนโดรเจน) ยังสามารถเริ่มต้นก่อนเวลาอันควร (adrenarche ก่อนกำหนด)
การตรวจคลื่นสมอง (คลื่นไฟฟ้าสมอง, EEG) ก็น่าสงสัยเช่นกัน
การตรวจทางพันธุกรรม
การทดสอบทางพันธุกรรมดำเนินการเพื่อยืนยันความสงสัยของกลุ่มอาการ Prader-Willi ในขั้นตอนแรก จะตรวจสอบการเกิดเมทิลเลชันของจุดสำคัญบนโครโมโซม 15 (15q11.2-q13, "SNRPN locus") เอ็นไซม์สามารถจับกลุ่มเมทิลที่เรียกว่ากับดีเอ็นเอและปรับเปลี่ยนได้ ในกรณีมากกว่า 99 เปอร์เซ็นต์ การตรวจนี้จะให้การวินิจฉัย มิฉะนั้น จะใช้วิธีทั่วไปอีกวิธีหนึ่งในการตรวจจับการเปลี่ยนแปลงของโครโมโซม คือ การเรืองแสงในแหล่งกำเนิดไฮบริไดเซชัน (FISH)
โรคที่คล้ายกันคือ Martin Bell Syndrome หรือ Angelmann Syndrome ความผิดปกติใน Martin Bell Syndrome อยู่บนโครโมโซม X (กลุ่มอาการ X ที่เปราะบาง) ในกรณีส่วนใหญ่ในกลุ่มอาการ Angelmann และกลุ่มอาการ Prader-Willi ตำแหน่งเดียวกันบนโครโมโซม 15 จะถูกลบออก แต่ในโรค Angelmann มีเพียงตำแหน่งเดียวบนโครโมโซมของมารดา
Prader-Willi Syndrome: การรักษา
ไม่มีวิธีรักษาโรค Prader-Willi อย่างไรก็ตาม ด้วยความช่วยเหลือของการบำบัดแบบประคับประคองที่ได้รับคำแนะนำอย่างเคร่งครัด อาการต่างๆ สามารถบรรเทาได้ องค์ประกอบหลักของการรักษาได้แก่ การควบคุมอาหาร การบำบัดด้วยฮอร์โมนทดแทน และการรักษาปัญหาด้านพฤติกรรม
สารอาหาร
ต้องได้รับสารอาหารที่เพียงพอและการเจริญเติบโตที่เพียงพอ โดยเฉพาะอย่างยิ่งหากกล้ามเนื้ออ่อนแรงรุนแรง สามารถใช้หัววัดหรือหัวนมเทียมแบบพิเศษเพื่ออำนวยความสะดวกในการรับประทานอาหาร นอกจากนี้ควรจัดทำแผนการให้อาหารที่มีปริมาณแคลอรี่และการควบคุมน้ำหนักที่บันทึกไว้เป็นอย่างดี
อย่างไรก็ตาม เมื่อเด็กโตขึ้น พวกเขาพัฒนาความผิดปกติของการกินมากเกินไป จากนั้นต้องปฏิบัติตามแผนที่มีการ จำกัด แคลอรี่อย่างเข้มงวด การจำกัดไขมันไม่ได้มุ่งเป้าเสมอไป เนื่องจากไขมันมีความสำคัญต่อการพัฒนาเขา ความผิดปกติของการกินอาจต้องมีการจัดการการเข้าถึงอาหารอย่างเข้มงวด ความผิดปกติของการกินในกลุ่มอาการ Prader-Willi มีผลกระทบร้ายแรงอย่างยิ่งต่อการเกิดโรค เนื่องจากผู้ที่ได้รับผลกระทบจะเคลื่อนไหวเพียงเล็กน้อย ดังนั้นจึงมีความแตกต่างกันมากระหว่างปริมาณแคลอรี่และความต้องการ จำเป็นอย่างยิ่งที่จะต้องให้ผู้ปกครองมีส่วนร่วมอย่างจริงจังและให้โครงสร้างที่มั่นคงแก่เด็กที่ป่วย
ในขณะเดียวกันก็ต้องมั่นใจว่ามีวิตามินและแร่ธาตุเพียงพอในกลุ่มอาการพราเดอร์-วิลลี่ ความผิดปกติของการเผาผลาญของกระดูกมักเกิดขึ้น ซึ่งสามารถป้องกันได้ด้วยวิตามินดีและการบริโภคแคลเซียม ควรติดตามพัฒนาการของโครงกระดูกโดยเฉพาะกระดูกสันหลัง
กิจกรรมและพัฒนาการทางการเคลื่อนไหวของเด็กป่วยจะได้รับการตรวจอย่างสม่ำเสมอ และหากจำเป็น ให้บำบัดรักษาด้วยกายภาพบำบัดหรือวิธีการที่คล้ายกัน
ฮอร์โมนการเจริญเติบโต
เมื่ออายุได้ 2 ขวบ ฮอร์โมนการเจริญเติบโต HGH สามารถให้เป็นยาได้ การรักษานี้ควรหยุดเมื่อปิดแผ่นการเจริญเติบโตในกระดูก (การควบคุม X-ray) การรักษาด้วยฮอร์โมนควรได้รับการตรวจสอบอย่างใกล้ชิดเสมอ การบริหารฮอร์โมนมีผลดีต่อการพัฒนาร่างกาย แต่ผลข้างเคียงคืออาการบวมน้ำที่เท้า ความโค้งของกระดูกสันหลัง (scoliosis) แย่ลง หรือความดันในกะโหลกศีรษะเพิ่มขึ้น (pseudotumor cerebri) ความผิดปกติของการหายใจอาจเกิดขึ้นได้เมื่อเริ่มการรักษา ดังนั้นจึงเป็นเรื่องสำคัญที่จะต้องติดตามการนอนหลับในช่วงเริ่มต้นของการรักษา การตรวจสอบการรักษาด้วยฮอร์โมน HGH ยังรวมถึงการตรวจวัดระดับไทรอยด์และระดับเลือดของปัจจัยการเจริญเติบโต IGF-1 ในเลือดอีกด้วย
ในกรณีของความผิดปกติของวัยแรกรุ่น ฮอร์โมนเพศสามารถให้การฉีดแบบดีโปต์ แผ่นแปะฮอร์โมน หรือเจล นี้สามารถปรับปรุงปัญหาพฤติกรรม เอสโตรเจนยังช่วยสร้างกระดูก แต่ก็มีผลข้างเคียงหลายประการ
การสนับสนุนทางจิตวิทยา
เด็กที่ได้รับผลกระทบควรได้รับความช่วยเหลือเพื่อสนับสนุนการพัฒนาพฤติกรรมและความสำเร็จของพัฒนาการที่สำคัญ ทักษะทางสังคมโดยเฉพาะอย่างยิ่งต้องได้รับการฝึกฝนในกลุ่มอาการพราเดอร์-วิลลี่ ควรส่งเสริมให้มีปฏิสัมพันธ์กับเพื่อนฝูงและผู้ดูแล อาจจำเป็นต้องมีการดูแลแบบตัวต่อตัวที่โรงเรียน หากจำเป็นจะต้องปรับสภาพแวดล้อมในการอยู่อาศัยและการทำงาน ความผิดปกติทางจิตเวชอาจต้องได้รับการบำบัดด้วยยา เช่น สารต้านเซโรโทนิน เป้าหมายของการสนับสนุนอย่างเข้มข้นคือความเป็นอิสระที่ดีที่สุด
อุปกรณ์ผ่าตัด
ปากแหว่งและเพดานโหว่ที่อาจมีอยู่แล้วตั้งแต่แรกเกิดสามารถรักษาได้โดยศัลยแพทย์ในระยะเริ่มแรก การวางแนวของดวงตาโดยเฉพาะอย่างยิ่งตำแหน่งเหล่สามารถรักษาได้ด้วยการผ่าตัดเพื่อป้องกันการรบกวนทางสายตา ประการแรก การปกปิดดวงตาที่แข็งแรงชั่วคราวสามารถช่วยได้
อวัยวะสืบพันธุ์ที่ด้อยพัฒนาอาจต้องได้รับการผ่าตัดเพื่อย้ายลูกอัณฑะจากช่องท้องส่วนล่างไปยังถุงอัณฑะ Beta-hCG (human chorionic gonadotropin) สามารถขยายถุงอัณฑะและทำให้อัณฑะจมได้
การเปลี่ยนแปลงของโครงกระดูกในกลุ่มอาการ Prader-Willi สามารถรักษาได้โดยการผ่าตัด ตำแหน่ง S ของกระดูกสันหลัง (scoliosis) จำเป็นต้องได้รับการผ่าตัดแก้ไขในกรณีที่รุนแรง แต่มักจะรักษาได้โดยไม่ต้องผ่าตัด (เช่น ใช้เครื่องรัดตัว
Prader-Willi syndrome: โรคและการพยากรณ์โรค
การวินิจฉัยโรค “Prader-Willi Syndrome” โดยเร็วที่สุดอาจส่งผลดีต่อการพยากรณ์โรคในระยะยาว อิทธิพลเชิงบวกต่อพฤติกรรม (การกิน) และการบริหารฮอร์โมนการเจริญเติบโตในที่สุดสามารถปรับปรุงคุณภาพชีวิตของเด็กที่ได้รับผลกระทบได้
ควรตรวจสอบพฤติกรรมการกิน น้ำหนัก และการเจริญเติบโตเป็นระยะหลังการวินิจฉัย มีการตรวจสอบพัฒนาการ การทำงาน พฤติกรรม ตลอดจนความผิดปกติทางจิตเวช การสังเกตและการดูแลอย่างใกล้ชิดโดยผู้เชี่ยวชาญที่สามารถประสานงานและดูแลสหวิทยาการเป็นสิ่งสำคัญมาก
คนป่วยมีความเสี่ยงที่จะติดเชื้อซ้ำๆ มากขึ้น ต้องใช้ความระมัดระวังเป็นพิเศษระหว่างการผ่าตัด: หลังจากการดมยาสลบ ระยะตื่นมักจะใช้เวลานานขึ้นและมีความเสี่ยงที่จะเกิดความผิดปกติของการหายใจเพิ่มขึ้น
อย่างไรก็ตาม ปัญหาที่ใหญ่ที่สุดคือการเพิ่มความอ้วน ในช่วงชีวิตภาวะแทรกซ้อนที่เกิดขึ้นมีอิทธิพลเหนือกว่า การตายก็เพิ่มขึ้นเนื่องจากภาวะแทรกซ้อน อัตราการเสียชีวิตที่เพิ่มขึ้นในกลุ่มอาการ Prader-Willi สาเหตุหลักมาจากโรคหัวใจ หลอดเลือดหรือปอด
ความเสี่ยงในการรับมรดกต่ำ ในกรณีส่วนใหญ่ การเปลี่ยนแปลงทางพันธุกรรมเกิดขึ้นเองตามธรรมชาติและไม่ได้รับการถ่ายทอด ความเสี่ยงสำหรับผู้ปกครองในการมีลูกคนที่สองที่มีอาการ Prader-Willi นั้นต่ำ ผู้ที่ได้รับผลกระทบมักจะไม่มีบุตร
เฉพาะในกรณีที่หายากเท่านั้นคือกลุ่มอาการ Prader-Willi ที่เกิดจากการเปลี่ยนแปลงข้อมูลทางพันธุกรรมในชุดโครโมโซมของผู้ปกครองซึ่งสืบทอดได้และยังนำไปสู่ความน่าจะเป็นในการสืบทอดที่สูงขึ้น หากเด็กมีอาการ Prader-Willi เราแนะนำให้ผู้ปกครองที่ต้องการมีลูกควรขอคำแนะนำจากนักพันธุศาสตร์ของมนุษย์
แท็ก: ฟิตเนส ค่าห้องปฏิบัติการ ข่าว