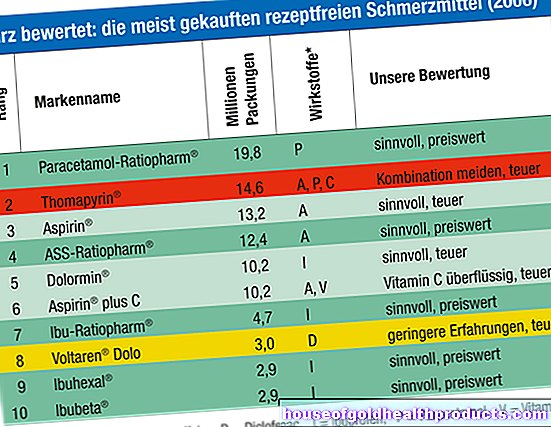
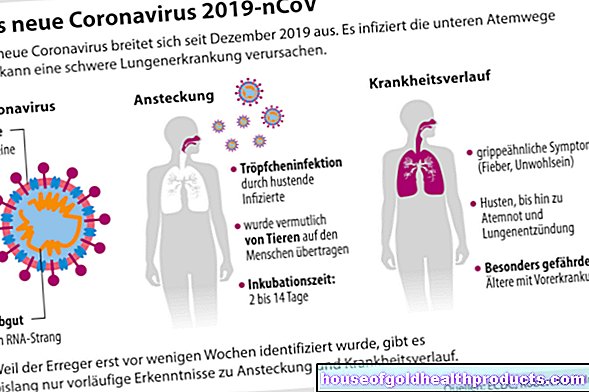
ฝ่ายกำกับดูแล
Martina Feichter ศึกษาวิชาชีววิทยาด้วยวิชาเลือกในร้านขายยาในเมือง Innsbruck และยังได้ดำดิ่งสู่โลกแห่งพืชสมุนไพรอีกด้วย จากที่นั่นก็ไม่ไกลจากหัวข้อทางการแพทย์อื่นๆ ที่ยังคงดึงดูดใจเธอมาจนถึงทุกวันนี้ เธอได้รับการฝึกฝนเป็นนักข่าวที่ Axel Springer Academy ในฮัมบูร์กและทำงานให้กับ มาตั้งแต่ปี 2550 โดยครั้งแรกในฐานะบรรณาธิการและตั้งแต่ปี 2555 เป็นนักเขียนอิสระ
ข้อมูลเพิ่มเติมเกี่ยวกับผู้เชี่ยวชาญของ เนื้อหา ทั้งหมดได้รับการตรวจสอบโดยนักข่าวทางการแพทย์
การค้นหาส่วนผสมออกฤทธิ์ใหม่ๆ เพื่อต่อต้านโรคหรืออาการป่วยบางอย่างเป็นเรื่องที่น่าเบื่อและไม่ได้จบลงด้วยความสำเร็จเสมอไป จากความหวัง 5,000 ถึง 10,000 รายที่ได้รับการทดสอบในห้องปฏิบัติการวิจัยของบริษัทยา โดยเฉลี่ยแล้วมีเพียงคนเดียวที่กลายเป็นยาสำเร็จรูปในร้านขายยา และมีค่าเฉลี่ย 13.5 ปีในระหว่าง

ค้นหา "เป้าหมาย"
แม้กระทั่งก่อนที่จะทำการทดสอบกับสารใหม่ นักวิจัยได้คำนึงถึงคุณสมบัติของสารที่เป็นปัญหาและปฏิกิริยาที่ควรกระตุ้นในร่างกาย เช่น การลดความดันโลหิต การปิดกั้นสารส่งสารบางชนิด หรือการปล่อยฮอร์โมน
ด้วยเหตุนี้ นักวิจัยจึงมองหา "เป้าหมาย" ที่เหมาะสม กล่าวคือ จุดโจมตีในกระบวนการเกิดโรค ซึ่งสารออกฤทธิ์สามารถยึดติดและส่งผลดีต่อกระบวนการเกิดโรค ในกรณีส่วนใหญ่ เป้าหมายคือเอนไซม์หรือตัวรับ (จุดเชื่อมต่อบนเซลล์สำหรับฮอร์โมนหรือสารส่งสารอื่นๆ) บางครั้งผู้ป่วยยังขาดสารบางอย่าง จากนั้นจะเห็นได้ชัดว่ายาที่คุณกำลังมองหาควรชดเชยข้อบกพร่องนี้อย่างรวดเร็ว ตัวอย่างที่รู้จักกันดีคืออินซูลินในผู้ป่วยเบาหวาน (เบาหวาน)
ค้นหาสารออกฤทธิ์
ทันทีที่มีการกำหนดเป้าหมาย นักวิทยาศาสตร์จะค้นหาสารออกฤทธิ์ที่สามารถทำหน้าที่ในการโจมตีที่เลือก (การตรวจคัดกรอง) ซึ่งมักจะหมายถึง: ทดสอบ ทดสอบ ทดสอบ มีการตรวจสอบสารต่างๆ มากถึง 300,000 ชนิดสำหรับความเหมาะสมทุกวัน (การคัดกรองปริมาณงานสูง = HTS) ในจำนวนนี้ ทุกๆ 200 ถึง 1000 ของสารเหล่านี้จะแสดงผลต่อเป้าหมายที่เลือกจริงๆ แม้ว่าบางครั้งจะเพียงเล็กน้อยก็ตาม การตีดังกล่าวเรียกว่า "การตี"
สารทดสอบส่วนใหญ่เป็นสารเคมี เช่น สังเคราะห์ขึ้น สารดัดแปลงพันธุกรรมก็ได้รับความสำคัญมาระยะหนึ่งแล้ว ได้มาจากความช่วยเหลือของเซลล์ดัดแปลงพันธุกรรม (เช่นแบคทีเรียบางชนิด) และสร้างพื้นฐานของชีวเภสัชภัณฑ์ (ยาชีวภาพ)
การเพิ่มประสิทธิภาพ
ในกรณีส่วนใหญ่ "Hit" ที่พบยังคงต้องได้รับการปรับให้เหมาะสม ตัวอย่างเช่น บางครั้ง ประสิทธิภาพของสารสามารถเพิ่มขึ้นได้หากโครงสร้างของสารเปลี่ยนแปลงเล็กน้อย ในการทดลองเหล่านี้ นักวิทยาศาสตร์มักจะทำงานกับการจำลองด้วยคอมพิวเตอร์ ซึ่งสามารถประเมินผลกระทบของการเปลี่ยนแปลงทางเคมีต่อสารได้ล่วงหน้า หากพยากรณ์โรคได้ดี สารจะถูกปรับในชีวิตจริง เช่น ในห้องปฏิบัติการ ผลกระทบต่อเป้าหมายจะถูกตรวจสอบอีกครั้ง
ด้วยวิธีนี้ นักวิจัยจะค่อยๆ ปรับปรุงสารออกฤทธิ์ใหม่ ซึ่งปกติจะใช้เวลาหลายปีในกรณีที่ดีที่สุด พวกเขาจะถึงจุดที่สารพร้อมสำหรับขั้นตอนต่อไป: มีการยื่นคำขอรับสิทธิบัตรแล้วจึงอยู่ภายใต้การศึกษาพรีคลินิกในฐานะผู้สมัครที่เรียกว่าสารออกฤทธิ์
พรีคลินิกศึกษา
ในระยะการพัฒนาพรีคลินิก (พรีคลินิก) ผู้สมัครยาได้รับการทดสอบในหลอดทดลอง (เช่น ในการเพาะเลี้ยงเซลล์) และในสัตว์ ในอีกด้านหนึ่ง เรื่องนี้เกี่ยวข้องกับปัญหาทางเภสัชวิทยา ตัวอย่างเช่น สิ่งที่เกิดขึ้นกับสารในเซลล์หรือในสิ่งมีชีวิตทั้งหมด:
- มันได้รับอย่างไร?
- กระจายในร่างกายอย่างไร?
- มันกระตุ้นปฏิกิริยาอะไร?
- จะดัดแปลงหรือรื้อถอน?
- เขาจะถูกกำจัดหรือไม่?
ในทางกลับกัน นักวิทยาศาสตร์กำลังตรวจสอบว่าสารมีผลกับเป้าหมายอย่างไร ระยะเวลานานเท่าใด และปริมาณเท่าใดที่จำเป็นสำหรับสารนั้น
อย่างไรก็ตาม เหนือสิ่งอื่นใด การศึกษาพรีคลินิกใช้เพื่อตอบคำถามเกี่ยวกับความเป็นพิษ (ความเป็นพิษ) ของตัวยา สารมีพิษหรือไม่? ทำให้เกิดมะเร็งได้หรือไม่? สามารถเปลี่ยนยีนได้หรือไม่? สามารถทำร้ายตัวอ่อนหรือทารกในครรภ์ได้หรือไม่?
ผู้สมัครยาจำนวนมากไม่ผ่านการทดสอบความเป็นพิษ เฉพาะสารที่ผ่านการทดสอบความปลอดภัยทั้งหมดเท่านั้นที่ได้รับอนุญาตให้เข้าสู่ขั้นตอนการพัฒนาต่อไปด้วยการศึกษาในมนุษย์ (การศึกษาทางคลินิก)
เมื่อใดก็ตามที่เป็นไปได้ การทดสอบพรีคลินิกจะดำเนินการในหลอดทดลอง เช่น ในการเพาะเลี้ยงเซลล์ ชิ้นส่วนของเซลล์ หรืออวัยวะของมนุษย์ที่แยกออกมาต่างหาก อย่างไรก็ตาม คำถามบางข้อสามารถตอบได้ในการทดสอบสิ่งมีชีวิตเท่านั้น และการทดลองกับสัตว์ก็เป็นสิ่งจำเป็นสำหรับสิ่งนี้
การศึกษาทางคลินิก
ผู้สมัครยากำลังถูกทดสอบกับมนุษย์เป็นครั้งแรกในการศึกษาทางคลินิก มีการแยกความแตกต่างระหว่างสามขั้นตอนการศึกษาที่สร้างจากกันและกัน:
- ระยะที่ 1: ผู้สมัครยาได้รับการทดสอบกับอาสาสมัครที่มีสุขภาพดีสองสามคน (กลุ่มทดสอบ)
- ระยะที่ II: ตามด้วยการทดสอบผู้ป่วยสองสามคน (เช่น ในผู้ป่วยความดันโลหิตสูงหากผู้ที่ได้รับยาจะกลายเป็นยาลดความดันโลหิตชนิดใหม่)
- ระยะที่ 3: ตอนนี้ทำการทดสอบกับผู้ป่วยจำนวนมาก
แต่ละขั้นตอนการศึกษาต้องได้รับการอนุมัติล่วงหน้าโดยหน่วยงานที่รับผิดชอบ: ด้านหนึ่ง ซึ่งรวมถึงหน่วยงานระดับชาติที่รับผิดชอบ - ขึ้นอยู่กับผู้สมัครยา ไม่ว่าจะเป็นสถาบันยาและอุปกรณ์การแพทย์แห่งสหพันธรัฐ (BfArM) หรือสถาบัน Paul Ehrlich (PEI) ). ในทางกลับกัน การศึกษาทางคลินิกทุกครั้งต้องได้รับอนุญาตจากคณะกรรมการจริยธรรม (ประกอบด้วยแพทย์ ทนายความ นักศาสนศาสตร์ และฆราวาส) ขั้นตอนนี้มีวัตถุประสงค์เพื่อปกป้องผู้เข้าร่วมการศึกษาในวิธีที่ดีที่สุด
ผู้ผลิตยาที่พัฒนาผู้สมัครยาสามารถดำเนินการศึกษาทางคลินิกด้วยตนเอง หรือเขาจ้าง "องค์กรวิจัยทางคลินิก" (CRO) ให้ทำเช่นนี้ เป็นบริษัทที่เชี่ยวชาญด้านการศึกษาทางคลินิก
การศึกษาระยะที่ 1
โดยปกติ ผู้ใหญ่ที่มีสุขภาพแข็งแรง 60 ถึง 80 คนที่อาสาทำหน้าที่นี้ในฐานะผู้ทดสอบในระยะที่ 1 หลังจากการอธิบายอย่างครอบคลุมและได้รับความยินยอมจากผู้เข้าร่วมการศึกษาวิจัยแล้ว ในขั้นต้นพวกเขาจะได้รับสารออกฤทธิ์เพียงเล็กน้อยเท่านั้น
ในการทดสอบต่อเนื่องกันสูงสุด 30 ครั้ง นักวิทยาศาสตร์ตรวจสอบว่าการค้นพบจากการทดสอบในหลอดทดลองและในสัตว์สามารถถ่ายทอดไปยังมนุษย์ได้หรือไม่ กล่าวคือ สารออกฤทธิ์ถูกดูดซึม กระจาย แปลง และขับออกมาอีกครั้งเช่นเดียวกับในพรีคลินิก กำหนดการทดสอบ นอกจากนี้ยังมีการตรวจสอบว่าผู้ทดสอบสามารถทนต่อยาได้ดีเพียงใด
แท็บเล็ตเข็มฉีดยาหรือครีม?
หลังจากผ่านช่วงที่ 1 สำเร็จแล้ว สิ่งที่เรียกว่ากาเลนิกส์ก็เข้ามามีบทบาท: ขณะนี้นักวิทยาศาสตร์กำลังทำงานเกี่ยวกับ "บรรจุภัณฑ์" ที่เหมาะสมที่สุดสำหรับสารออกฤทธิ์ - ควรฉีดเข้าเส้นเลือดเป็นยาเม็ด แคปซูล ยาเหน็บ เข็มฉีดยา หรือยาฉีดหรือไม่?
คำตอบสำหรับคำถามนี้มีความสำคัญมาก: รูปแบบของขนาดยามีอิทธิพลอย่างมากต่อความน่าเชื่อถือ ความรวดเร็วและระยะเวลาที่สารออกฤทธิ์จะทำหน้าที่ในร่างกายให้สำเร็จ นอกจากนี้ยังส่งผลต่อประเภทและความรุนแรงของผลข้างเคียงที่อาจเกิดขึ้น สารออกฤทธิ์บางชนิดสามารถทนต่อการฉีดได้ดีกว่าเมื่อเข้าสู่ร่างกายในรูปแบบแท็บเล็ตผ่านทางทางเดินอาหาร
นอกจากนี้ ผู้เชี่ยวชาญทางกาลินาจะตรวจสอบว่าควรเติมสารเสริมชนิดใดในการเตรียมการใหม่หรือไม่ ตัวอย่างเช่น สิ่งที่ช่วยเพิ่มรสชาติของยาหรือทำหน้าที่เป็นพาหะหรือสารกันบูด
คุณสามารถอ่านเพิ่มเติมเกี่ยวกับการค้นหา "บรรจุภัณฑ์" ที่ถูกต้องสำหรับสารออกฤทธิ์ใหม่และวัสดุเสริมที่เหมาะสมได้ในบทความ Galenics - การผลิตยา
การศึกษาระยะที่ 2 และระยะที่ 3
หลังจากอาสาสมัครที่มีสุขภาพดีในระยะที่ 1 จะถึงคราวของผู้ป่วยจากระยะที่ 2 เพื่อทดสอบตัวรับยา:
- ระยะที่ 2 : ในที่นี้ ตัวยาใหม่ได้รับการทดสอบในผู้ป่วย 100 ถึง 500 รายเป็นส่วนใหญ่ เน้นที่ประสิทธิภาพ ปริมาณที่เหมาะสม และความทนทานของการเตรียมการ
- ระยะที่ 3: การตรวจสอบแบบเดียวกันดำเนินการที่นี่เช่นเดียวกับในระยะที่ 2 เฉพาะกับผู้ป่วยจำนวนมากขึ้นอย่างมีนัยสำคัญ (หลายพันคน) นอกจากนี้ยังให้ความสนใจกับการโต้ตอบที่เป็นไปได้กับยาอื่น ๆ
ในทั้งสองขั้นตอน มีการเปรียบเทียบการรักษาที่แตกต่างกัน: ผู้ป่วยบางรายเท่านั้นที่ได้รับการเตรียมการใหม่ ส่วนที่เหลือจะได้รับยามาตรฐานปกติหรือที่คุ้นเคยหรือยาหลอก - การเตรียมการที่ดูเหมือนใหม่ แต่ไม่มีสาร สารออกฤทธิ์ (ยาหลอก) . ตามกฎแล้วทั้งผู้ป่วยและแพทย์ที่รักษาไม่ทราบว่าใครได้รับอะไร "การศึกษาแบบ double-blind" ดังกล่าวได้รับการออกแบบมาเพื่อป้องกันความหวัง ความกลัว หรือทัศนคติที่สงสัยของแพทย์และผู้ป่วยจากอิทธิพลของผลลัพธ์ของการรักษา
กำลังอนุมัติ
แม้ว่ายาตัวใหม่จะผ่านการศึกษาและการทดสอบที่จำเป็นทั้งหมดแล้ว แต่ก็ไม่สามารถขายได้ ในการดำเนินการดังกล่าว บริษัทยาต้องยื่นขออนุมัติยาจากหน่วยงานที่มีอำนาจก่อน (ดูด้านล่าง: ตัวเลือกการอนุมัติ) สิ่งนี้จะตรวจสอบผลการศึกษาทั้งหมดอย่างรอบคอบ และในกรณีที่ดีที่สุด อนุญาตให้ผู้ผลิตนำยาใหม่ออกสู่ตลาด
ระยะที่สี่
แม้ว่ายาจะได้รับการอนุมัติแล้ว ทางการและบริษัทยาก็จับตาดูการเตรียมการใหม่ เช่น เกี่ยวกับผลข้างเคียงที่หายาก สิ่งเหล่านี้เป็นผลที่ไม่พึงประสงค์ที่เกิดขึ้นในผู้ป่วยที่ได้รับการรักษาน้อยกว่า 1 ใน 10,000 ดังนั้นจึงแทบจะไม่สามารถตรวจพบได้ในระยะการศึกษาก่อนหน้านี้ (กับกลุ่มผู้ป่วยที่เล็กกว่า) แพทย์จำเป็นต้องรายงานผลข้างเคียงที่ไม่คาดคิดของยา
หากจำเป็น หน่วยงานอนุมัติจะขอให้ผู้ผลิตชี้ให้เห็นผลข้างเคียงที่ค้นพบใหม่เหล่านี้ในส่วนแทรกของบรรจุภัณฑ์ อย่างไรก็ตาม ยานี้ยังสามารถออกข้อจำกัดในการใช้งานได้ เช่น หากพบผลข้างเคียงที่หายากแต่รุนแรงในบริเวณไต ทางการสามารถสั่งไม่ให้ใช้ยานี้กับผู้ที่เป็นโรคไตอยู่แล้วได้
ในกรณีร้ายแรง เจ้าหน้าที่สามารถเพิกถอนการอนุมัติยาได้ทั้งหมด หากเมื่อเวลาผ่านไปมีความเสี่ยงที่ยอมรับไม่ได้จากการใช้ยา บางครั้งผู้ผลิตก็ถอนผลิตภัณฑ์ดังกล่าวออกจากตลาดโดยสมัครใจ
แพทย์ยังใช้บันทึกเพื่อบันทึกว่ายาตัวใหม่นี้ทำหน้าที่ในชีวิตประจำวันของผู้ป่วยอย่างไร ผู้ผลิตใช้ผลของการศึกษาเชิงสังเกตดังกล่าว ตัวอย่างเช่น เพื่อปรับปรุงขนาดยาหรือรูปแบบขนาดการใช้ของยาเตรียม
บางครั้งการปฏิบัติในชีวิตประจำวันยังแสดงให้เห็นว่าสารออกฤทธิ์ช่วยต่อต้านโรคอื่นๆ ผู้ผลิตมักจะทำการวิจัยต่อไปในทิศทางนี้ ด้วยการศึกษาระยะที่ II และ III ใหม่ หากสำเร็จ เขาสามารถยื่นขออนุมัติสิ่งบ่งชี้ใหม่นี้ได้
ตัวเลือกการอนุมัติ
โดยหลักการแล้ว บริษัทยาสามารถยื่นขออนุมัติยาใหม่ได้สำหรับทั้งสหภาพยุโรปหรือประเทศสมาชิกเพียงประเทศเดียว:
กระบวนการอนุมัติจากส่วนกลาง
ขอการอนุมัติยาได้ที่นี่โดยตรงจาก European Medicines Agency (EMA) หน่วยงานอนุมัติของประเทศสมาชิกสหภาพยุโรปก็มีส่วนร่วมในการทดสอบครั้งต่อไปเช่นกัน หากใบสมัครได้รับการอนุมัติ การเตรียมการสามารถขายได้ทุกที่ในสหภาพยุโรป กระบวนการอนุมัตินี้ใช้เวลาเฉลี่ย 1 ปีครึ่ง และจำเป็นสำหรับยาบางชนิด (เช่น สำหรับยาที่ผลิตด้วยเทคโนโลยีชีวภาพและสำหรับยารักษามะเร็งที่มีส่วนผสมออกฤทธิ์ใหม่)
ขั้นตอนการอนุมัติระดับประเทศ
คำขออนุมัติจะถูกส่งไปยังหน่วยงานระดับประเทศและเฉพาะในประเทศที่เกี่ยวข้องเท่านั้น ในเยอรมนี Federal Institute for Drugs and Medical Devices (BfArM) และ Paul Ehrlich Institute (PEI) มีหน้าที่รับผิดชอบในเรื่องนี้ BfArM ดูแลเภสัชภัณฑ์ของมนุษย์ส่วนใหญ่ PEI ดูแลซีรั่ม วัคซีน สารก่อภูมิแพ้ในการทดสอบ ซีรั่มทดสอบและแอนติเจนทดสอบ เลือดและผลิตภัณฑ์จากเลือด เนื้อเยื่อและยาสำหรับยีนบำบัดและการบำบัดด้วยเซลล์
การอนุมัติยาในหลายประเทศในสหภาพยุโรป
นอกจากนี้ยังมีอีกสองทางเลือกหากบริษัทยาต้องการขออนุมัติในหลายประเทศในสหภาพยุโรป:
- ขั้นตอนการกระจายอำนาจ: ใน "ขั้นตอนการกระจายอำนาจ" (DCP) บริษัทยาสามารถยื่นขอการอนุมัติระดับชาติสำหรับยาใหม่ในหลายประเทศของเขตเศรษฐกิจยุโรปในเวลาเดียวกัน
- กระบวนการรับรู้ร่วมกัน: หากยาได้รับการอนุมัติระดับประเทศแล้วในประเทศในเขตเศรษฐกิจยุโรป รัฐสมาชิกอื่น ๆ สามารถรับรู้สิ่งนี้ได้ภายในกรอบของ "ขั้นตอนการรับรู้ร่วมกัน" (MRP)
การขออนุมัติยาใหม่มีราคาแพงมากสำหรับบริษัทยา ตัวอย่างเช่น การประมวลผลคำร้องเพื่อขออนุมัติส่วนผสมออกฤทธิ์ใหม่ที่ EMA มีค่าใช้จ่ายประมาณ 260,000 ยูโร ในกรณีที่ง่ายที่สุด
มาตรฐานการรับรอง
ยาบางชนิดออกจำหน่ายโดยได้รับอนุมัติมาตรฐาน: ยาเหล่านี้ไม่ใช่ยาที่พัฒนาขึ้นใหม่ แต่เป็นยาที่ผลิตขึ้นจากเอกสารที่สภานิติบัญญัติกำหนด นอกจากนี้ ยาเหล่านี้ต้องไม่เป็นอันตรายต่อมนุษย์หรือสัตว์ ในเอกสาร (เช่น สำหรับยาเหน็บพาราเซตามอล 250 มก.) เหนือสิ่งอื่นใด องค์ประกอบและปริมาณของยาที่เป็นปัญหานั้นถูกกำหนดไว้อย่างแม่นยำ - เช่นเดียวกับพื้นที่ของแอปพลิเคชัน
หากเป็นไปตามข้อกำหนดทั้งหมด ผู้ผลิตไม่จำเป็นต้องขออนุมัติยาเป็นรายบุคคล ทำให้เขาสามารถนำยาออกสู่ตลาดได้ในราคาที่ไม่แพงมาก มีการอนุมัติมาตรฐานสำหรับเม็ดถ่าน (250 มก.) ยาหยอดตา atropine และสารละลายที่มีความเข้มข้นต่างๆ รวมทั้งยาเหน็บพาราเซตามอลและยาเม็ดกรดอะซิติลซาลิไซลิกในปริมาณต่างๆ
ตัวอย่างเช่น เภสัชกรได้รับอนุญาตให้เตรียมน้ำเกลือตามคำแนะนำในตำรับยาที่เกี่ยวข้องแล้วขาย อย่างไรก็ตาม คุณต้องระบุการใช้การอนุมัติมาตรฐานดังกล่าวต่อหน่วยงานอนุมัติและหน่วยงานของรัฐที่รับผิดชอบ
วิธีอื่นในการขออนุมัติยา
ในสหภาพยุโรป นอกเหนือจากขั้นตอนการอนุมัติตามแบบแผนแล้ว ยังมีทางเลือกสำหรับการผลิตยาตัวใหม่เร็วกว่าปกติอีกด้วย สิ่งเหล่านี้ไม่ได้เป็นเพียงการอนุมัติอย่างรวดเร็ว ในทางกลับกัน มีความพยายามหลายวิธีเพื่อให้แน่ใจว่าผู้ที่ได้รับผลกระทบจะได้รับประโยชน์จากส่วนผสมออกฤทธิ์ แม้จะไม่มีการอนุมัติยาแผนโบราณก็ตาม ผู้เชี่ยวชาญพูดถึงเส้นทางการปรับตัวที่เรียกว่า:
โปรแกรมใช้งานอย่างเห็นอกเห็นใจ
ที่นี่ ผู้ป่วยที่เฉพาะเจาะจงมาก ๆ จะได้รับยาที่จริง ๆ แล้วยังอยู่ในระหว่างการทดลองทางคลินิก ข้อกำหนดเบื้องต้นคือไม่มีทางเลือกในการรักษาอื่นๆ อีกต่อไป และผู้ป่วยไม่สามารถเข้าร่วมในการศึกษาที่สอดคล้องกันเกี่ยวกับยานี้ ต้องใช้ข้อยกเว้นเหล่านี้สำหรับผู้ป่วยแต่ละรายแยกกัน
การอนุมัติตามเงื่อนไขสำหรับผลิตภัณฑ์ยา
นี่คือการอนุมัติอย่างรวดเร็ว ไม่จำเป็นต้องมีการทดสอบประสิทธิภาพและความปลอดภัยที่เข้มงวดเท่าที่ปกติ อย่างไรก็ตาม มีเงื่อนไขบางประการดังต่อไปนี้:
- การอนุมัติยาตามเงื่อนไขมีเวลาจำกัด
- ผู้ผลิตต้องจัดเตรียมเอกสารที่ขาดหายไปที่จำเป็นสำหรับการอนุมัติยาเป็นประจำ
การอนุมัติแบบมีเงื่อนไขจะใช้ ตัวอย่างเช่น ในการระบาดใหญ่เพื่อจัดหายาที่เหมาะสมกับโรคติดเชื้อได้อย่างรวดเร็ว
การอนุมัติภายใต้สถานการณ์พิเศษ
เส้นทางพิเศษนี้มีให้ เช่น โรคหายาก เนื่องจากมีคนป่วยน้อยมาก บริษัทยาจึงไม่สามารถส่งข้อมูลจำนวนที่จำเป็นไปตรวจสอบได้ อย่างไรก็ตาม ด้วยการอนุมัติยานี้ ผู้ผลิตมักจะต้องตรวจสอบทุกปีว่ามีข้อมูลและข้อค้นพบใหม่หรือไม่
การอนุมัติยาแบบเร่งรัด (การประเมินแบบเร่งรัด)
เอกสารการอนุมัติจะได้รับการตรวจสอบและประเมินผลเร็วขึ้นโดยคณะกรรมการ EMA ที่รับผิดชอบ - แทนที่จะเป็น 210 ปกติใน 150 วัน เส้นทางนี้เป็นไปได้หากมีสารออกฤทธิ์ที่มีแนวโน้มว่าจะต่อต้านโรคที่ไม่สามารถรักษาได้อย่างถูกต้องจนถึงปัจจุบัน
ยาสำคัญ (PRIME)
ในกรณีดังกล่าวที่ยังคงไม่ตอบสนองความต้องการ EMA และผู้ผลิตยาสามารถทำงานร่วมกันได้ตั้งแต่เนิ่นๆ แม้กระทั่งในระหว่างการทดสอบครั้งแรก ด้วยวิธีนี้ ผู้เชี่ยวชาญสามารถประเมินประสิทธิภาพและความปลอดภัยได้ตั้งแต่ระยะเริ่มต้น และเริ่มขั้นตอนเพิ่มเติมได้รวดเร็วยิ่งขึ้นหากยาพิสูจน์แล้วว่ามีแนวโน้มดี
การตรวจสอบอย่างต่อเนื่อง (การตรวจสอบแบบต่อเนื่อง)
ในกรณีของยาและวัคซีนที่จำเป็นอย่างเร่งด่วน EMA สามารถ "ตามเงื่อนไข" อนุมัติส่วนผสมออกฤทธิ์หรือทำงานร่วมกับผู้ผลิตในระยะแรกก่อนที่จะได้รับการอนุมัติขั้นสุดท้าย ในกรณีที่สำคัญ กระบวนการตรวจสอบแบบต่อเนื่องที่เรียกว่าเริ่มต้นก่อนการอนุมัติเหล่านี้ ผู้เชี่ยวชาญจะประเมินข้อมูลที่มีอยู่ก่อนที่ผู้ผลิตจะสามารถส่งเอกสารทั้งหมดที่เกี่ยวข้องเพื่อขออนุมัติได้ นอกจากนี้ พวกเขายังตรวจสอบผลลัพธ์ใหม่ทั้งหมดที่ได้รับจากการศึกษาเพิ่มเติมอย่างต่อเนื่อง
ตัวอย่างเช่น EMA นำกระบวนการตรวจสอบแบบกลิ้งไปใช้กับการอนุมัติตามเงื่อนไขของยาเรมเดซิเวียร์ที่ติดไวรัสระหว่างการระบาดของโคโรนาไวรัส ในกระบวนการอนุมัติวัคซีนโคโรนา ผู้เชี่ยวชาญยังได้ตรวจสอบผลลัพธ์ที่มีอยู่แล้วและได้รับในระหว่างการศึกษาระยะที่ 3 ที่กำลังดำเนินอยู่
ยาสำหรับเด็ก
ยาชนิดใหม่มักจะต้องผ่านการศึกษาหลายชิ้นก่อนจะได้รับอนุญาตให้ออกสู่ตลาด อย่างไรก็ตาม เป็นเวลานานแล้วที่ผู้ป่วยกลุ่มหนึ่งได้รับความสนใจน้อยลงในการวิจัย ทั้งเด็กและวัยรุ่น สำหรับการรักษาผู้เยาว์ ปริมาณของยาที่ได้รับการทดสอบในผู้ใหญ่มักจะลดลงอย่างง่ายๆ
อย่างไรก็ตาม ตั้งแต่ปี 2550 ยาใหม่ทุกตัวในสหภาพยุโรปต้องได้รับการทดสอบกับผู้เยาว์ในการศึกษาระยะที่ II และ III หากต้องใช้ในกลุ่มอายุนี้ในภายหลัง การทดสอบกับเด็กหรือวัยรุ่นมักเริ่มต้นเมื่อการศึกษาระยะที่ 2 เกี่ยวกับผู้ใหญ่เสร็จเรียบร้อยแล้วเท่านั้น กลุ่มผู้เชี่ยวชาญแยกต่างหากจาก European Medicines Agency EMA ซึ่งเป็นคณะกรรมการกุมารเวชศาสตร์ ตัดสินใจในรายละเอียด
การทดสอบการรับเข้าเรียนในผู้เยาว์นั้นสมเหตุสมผลเพราะร่างกายของเด็กและวัยรุ่นมักตอบสนองต่อยาที่แตกต่างจากของผู้ใหญ่ ประสิทธิผลและความทนทานจึงแตกต่างกัน ดังนั้นจึงต้องปรับขนาดยาสำหรับผู้เยาว์ ในหลายกรณี การใช้ยาสำหรับเด็กต้องใช้รูปแบบอื่น เช่น ยาหยอดหรือผงแทนยาเม็ดขนาดใหญ่ที่ผู้ป่วยผู้ใหญ่ได้รับ
ยาสมุนไพร
เมื่อพัฒนาผลิตภัณฑ์ยาสมุนไพรใหม่ (ยารักษาโรคพืช) การพิสูจน์ประสิทธิภาพตามที่กำหนดในรูปแบบของการศึกษาทางคลินิกเป็นเรื่องยาก:
แม้ว่ายาเคมีมักจะมีสารบริสุทธิ์ไม่เกินหนึ่งหรือสองชนิด แต่พืชแต่ละชนิดก็ผลิตสารออกฤทธิ์ผสม โดยส่วนใหญ่แล้ว ส่วนผสมนี้จะแตกต่างกันไปตามส่วนต่างๆ ของพืช ตัวอย่างเช่น สมุนไพรตำแยอาจส่งผลต่อไต ในขณะที่รากตำแยอาจส่งผลต่อการเผาผลาญฮอร์โมนของต่อมลูกหมาก นอกจากนี้ ส่วนผสมของสารออกฤทธิ์เหล่านี้จะแตกต่างกันไปตามแหล่งกำเนิดและการเตรียมพืช ซึ่งส่งผลต่อประสิทธิภาพด้วยเช่นกัน
ในปี 1978 กลุ่มผู้เชี่ยวชาญที่เรียกว่า Commission E ได้ถูกจัดตั้งขึ้นเพื่อชี้แจงคำถามดังกล่าว ข้อมูลเหล่านี้ประกอบด้วยข้อมูลที่ทราบในขณะนั้นเกี่ยวกับองค์ประกอบ ผลกระทบ และผลข้างเคียงที่เป็นไปได้ของพืชสมุนไพรต่างๆ
เนื่องจากเอกสารของ Commission E ไม่ได้รับการปรับปรุงตั้งแต่ปี 1994 จึงใช้เอกสารของ "คณะกรรมการผลิตภัณฑ์ยาสมุนไพร" (HMPC) แทน นี่คือคณะกรรมการของ European Medicines Agency ที่รับผิดชอบด้านยาสมุนไพร เขาดูแลการประเมินทางวิทยาศาสตร์ของยาดังกล่าว
ต้องแยกความแตกต่างระหว่างผลิตภัณฑ์ยาสมุนไพรแบบดั้งเดิมกับผลิตภัณฑ์ยาสมุนไพรสมัยใหม่: แทนที่จะต้องได้รับการอนุมัติ จำเป็นต้องลงทะเบียน เพิ่มเติมเกี่ยวกับเรื่องนี้ในหัวข้อถัดไป
การลงทะเบียนแทนการรับเข้าเรียน
ผลิตภัณฑ์ยาจากสมุนไพรแบบดั้งเดิมและยาชีวจิตได้รับการยกเว้นจากข้อกำหนดด้านใบอนุญาตว่าเป็นผลิตภัณฑ์ยา "การบำบัดพิเศษ" คุณต้องลงทะเบียนแทน:
สำหรับสิ่งนี้ - ด้วยการอนุมัติผลิตภัณฑ์ยา "ปกติ" - ต้องส่งหลักฐานการไม่เป็นอันตรายและคุณภาพยาที่เหมาะสมของผลิตภัณฑ์ยาชีวจิตหรือสมุนไพรดั้งเดิม
ในกรณีของผลิตภัณฑ์ยาสมุนไพรแผนโบราณ จะต้องแสดงให้เห็นผลทางเภสัชวิทยาหรือประสิทธิผลอย่างสมเหตุสมผล โดยใช้สิ่งที่เรียกว่าหลักฐานดั้งเดิม ซึ่งหมายความว่าผู้ผลิตต้องใช้ข้อมูลบรรณานุกรมเพื่อพิสูจน์ว่าผลิตภัณฑ์ยาสมุนไพรแบบดั้งเดิมมีการใช้ทางการแพทย์ในสหภาพยุโรปอย่างน้อย 30 ปีรวมถึงอย่างน้อย 15 ปี
อย่างไรก็ตาม การศึกษาทางคลินิกเพื่อพิสูจน์ประสิทธิภาพ ตามที่กำหนดโดยการอนุมัติยาแผนโบราณนั้น ไม่จำเป็นสำหรับยาชีวจิตหรือยาสมุนไพรแผนโบราณ ดังนั้นบริษัทจึงสามารถขายได้
ตรงกันข้ามกับยาแผนโบราณในการแพทย์แผนโบราณ การเยียวยาทางเลือกมักจะไม่มีหลักฐานทางวิทยาศาสตร์ที่กว้างขวางถึงประสิทธิผลของยา โดยเฉพาะอย่างยิ่งเนื่องจากไม่ต้องผ่านกระบวนการอนุมัติยาที่ใช้เวลานาน
แท็ก: ตั้งครรภ์ สุขภาพของผู้หญิง อยากมีบุตร

.jpg)