ฟีนิลคีโตนูเรีย
Marian Grosser ศึกษาการแพทย์ของมนุษย์ในมิวนิก นอกจากนี้ แพทย์ผู้สนใจในหลายๆ สิ่ง กล้าที่จะออกนอกเส้นทางที่น่าตื่นเต้น เช่น ศึกษาปรัชญาและประวัติศาสตร์ศิลปะ ทำงานทางวิทยุ และสุดท้ายก็เพื่อ Netdoctor ด้วย
ข้อมูลเพิ่มเติมเกี่ยวกับผู้เชี่ยวชาญของ เนื้อหา ทั้งหมดได้รับการตรวจสอบโดยนักข่าวทางการแพทย์Phenylketonuria (PKU) เป็นโรคทางพันธุกรรมที่มีมา แต่กำเนิดของการเผาผลาญโปรตีน ช่วยป้องกันการสลายตัวของกรดอะมิโนฟีนิลอะลานีน สิ่งนี้สะสมในร่างกายและขัดขวางการพัฒนาสมองของเด็ก หากไม่ได้รับการรักษา ฟีนิลคีโตนูเรียจะนำไปสู่ความบกพร่องทางสติปัญญาขั้นรุนแรง อย่างไรก็ตาม ด้วยการรักษาอย่างทันท่วงที ผู้ป่วยสามารถมีชีวิตที่ปกติได้ ค้นหาทุกสิ่งเกี่ยวกับฟีนิลคีโตนูเรียได้ที่นี่!
รหัส ICD สำหรับโรคนี้: รหัส ICD เป็นรหัสที่เป็นที่ยอมรับในระดับสากลสำหรับการวินิจฉัยทางการแพทย์ สามารถพบได้เช่นในจดหมายของแพทย์หรือในใบรับรองความสามารถในการทำงาน E70
ฟีนิลคีโตนูเรีย: คำอธิบาย
Phenylketonuria (PKU) เป็นโรคเมตาบอลิซึมที่สืบทอดมาซึ่งเกิดขึ้นตั้งแต่แรกเกิดและรบกวนการสลายของฟีนิลอะลานีนกรดอะมิโนที่จำเป็น กรดอะมิโนเป็นส่วนประกอบพื้นฐานของโปรตีนและเป็นส่วนประกอบที่สำคัญในการเผาผลาญ บางชนิดสามารถเข้าสู่ร่างกายได้ทางอาหารเท่านั้น ร่างกายไม่สามารถผลิตเองได้ กรดอะมิโนดังกล่าวเรียกว่าจำเป็น
เกิดอะไรขึ้นกับฟีนิลคีโตนูเรีย?
โดยปกติกรดอะมิโนจะมีความสมดุลระหว่างการดูดซึม / การสะสมและการสลายเพื่อให้มีมากที่สุดเท่าที่ร่างกายต้องการ ในกรณีของกรดอะมิโนหลายชนิด การขาดหรือมากเกินไปอาจก่อให้เกิดอันตรายอย่างมากและทำให้เกิดอาการต่างๆ
ใน PKU แบบคลาสสิกที่เรียกว่า ผลของฟีนิลอะลานีนไฮดรอกซีเลส (PAH) นั้นจำกัดหรือขาดหายไปโดยสิ้นเชิง เนื่องจากการขาด PAH ฟีนิลอะลานีนจึงสะสมในร่างกายมากขึ้น ความเข้มข้นของฟีนิลอะลานีนที่สูงเกินไปจะขัดขวางการพัฒนาของสมองอย่างมาก และนำไปสู่ความบกพร่องทางสติปัญญาในผู้ป่วยอายุน้อยในระยะเริ่มแรก
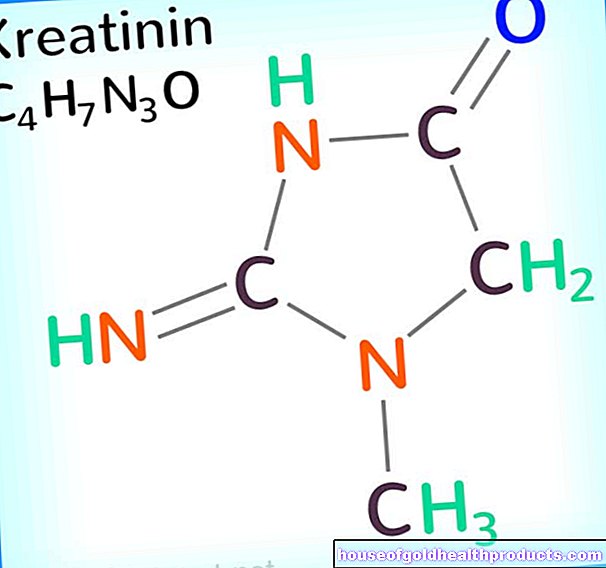
เนื่องจากโรคนี้ไม่สามารถสลายฟีนิลอะลานีนตามปกติได้ จึงเกิดผลิตภัณฑ์จากการสลายอื่น ๆ ที่เรียกว่าฟีนิลคีโตนขึ้น พวกมันถูกขับออกทางปัสสาวะและรับผิดชอบต่อชื่อของโรค
ฟีนิลคีโตนูเรียผิดปกติ
แม้จะมีรูปแบบที่ผิดปรกติของฟีนิลคีโตนูเรีย การสลายของฟีนิลอะลานีนก็ยังถูกรบกวน อย่างไรก็ตาม สาเหตุไม่ใช่ข้อบกพร่องใน PAH แต่กลับถูกจำกัดการทำงานของโคเอ็นไซม์ tetrahydrobiopterin (BH4) มีส่วนเกี่ยวข้องทางอ้อมในการสลายตัวของฟีนิลอะลานีนเนื่องจาก PAH ต้องการ BH4 เพื่อเปลี่ยนฟีนิลอะลานีนเป็นไทโรซีน
เนื่องจาก BH4 มีความสำคัญต่อการผลิตสาร dopamine และ serotonin สารส่งสาร phenylketonuria ที่ผิดปกติโดยขาด BH4 มักจะซับซ้อนกว่ารูปแบบคลาสสิก
ฟีนิลคีโตนูเรียส่งผลต่อใครบ้าง?
PKU เป็นหนึ่งในโรคเมแทบอลิซึมที่มีมา แต่กำเนิดที่พบบ่อยที่สุด คาดว่าประมาณหนึ่งใน 7,000 ของทารกแรกเกิดทั่วโลกจะพัฒนาโดยไม่มีความแตกต่างระหว่างเด็กหญิงและเด็กชาย เนื่องจากเป็นโรคทางพันธุกรรม สมาชิกในครอบครัวหลายคนจึงมักได้รับผลกระทบ
Phenyketonuria: อาการ
ในตอนแรก ทารกที่มีฟีนิลคีโตนูเรียไม่แสดงอาการของโรค ปัญหาแรกจะไม่เกิดขึ้นจนกว่าจะถึงเดือนที่สี่ถึงเดือนที่หกของชีวิตหากโรคยังไม่ได้รับการยอมรับและรักษา เหนือสิ่งอื่นใด การเจริญเติบโตของสมองที่ถูกรบกวนทำให้เกิดภาวะแทรกซ้อนครั้งใหญ่เมื่อเวลาผ่านไป อาการของ PKU ที่ไม่ได้รับการรักษา ได้แก่:
- การขาดดุลทางจิตที่แข็งแกร่ง ความเสียหายของสมองดำเนินไปตามวัยแรกรุ่นและซบเซา เด็กที่ได้รับผลกระทบมักมีความพิการทางจิตใจอย่างรุนแรง
- พอดี (ชัก) (โรคลมชัก). เนื่องจากความเสียหาย เซลล์ประสาทของสมองจึงมีความอ่อนไหวเป็นพิเศษและสามารถกระตุ้นได้มากเกินไป อาการชักจากโรคลมชักบ่อยครั้งเป็นผล
- ความพิการของมอเตอร์ ไม่เพียงแต่เซลล์สมองเท่านั้นแต่ยังทำให้กล้ามเนื้อของผู้ป่วยตื่นเต้นมากเกินไปอีกด้วย นั่นคือเหตุผลที่มักจะเกร็ง (เกร็ง) ซึ่งนำไปสู่ความผิดปกติของการเคลื่อนไหวต่างๆ
- พฤติกรรมผิดปกติ เด็กบางคนที่มีฟีนิลคีโตนูเรียมีสมาธิสั้นและก้าวร้าวผิดปกติ และความโกรธก็เกิดขึ้นได้บ่อยเช่นกัน
- หัวเล็ก (microcephaly) เนื่องจากสมองของผู้ป่วยไม่พัฒนาอย่างเหมาะสม การเจริญเติบโตของศีรษะก็ลดลงเช่นกัน เส้นรอบวงศีรษะที่เล็กเมื่อเทียบกับคนรอบข้างจะสังเกตเห็นได้ชัดเจนในเด็กโต
- กลิ่นที่เห็นได้ชัดเจน PKU ผลิตผลิตภัณฑ์ที่สลายตัวของฟีนิลอะลานีนซึ่งมีกลิ่นคล้ายกับอุจจาระของหนู สารเหล่านี้ส่วนใหญ่ถูกขับออกทางปัสสาวะ แต่ยังบางส่วนผ่านทางผิวหนัง
- การเปลี่ยนแปลงของผิวหนังเหมือนกลาก
เนื่องจากการผลิตเม็ดสีเมลานินยังถูกรบกวนในฟีนิลคีโตนูเรีย ผู้ประสบภัยจำนวนมากจึงมีผิวที่บอบบาง แพ้แดด และผมสีบลอนด์ขาว ม่านตายังเป็นสีฟ้าอ่อนถึงโปร่งใส และช่วยให้อวัยวะที่เป็นสีแดงส่องผ่านได้
อาการของ PKU แตกต่างกันไปตามความรุนแรงในแต่ละคน สาเหตุหลักคือกิจกรรมของ phenylalanine hydroxylase (PAH) ถูกจำกัดในผู้ป่วยแต่ละรายแตกต่างกัน บางคนยังคงมีกิจกรรมตกค้างบางอย่างเพื่อให้ฟีนิลอะลานีนสะสมในร่างกายน้อยลง บางส่วนไม่แสดงกิจกรรมของเอนไซม์เลย - โรคดำเนินไปอย่างรวดเร็วและจริงจังมากขึ้นตามลำดับ
ฟีนิลคีโตนูเรีย: สาเหตุและปัจจัยเสี่ยง
Phenylketonuria เป็นโรคทางพันธุกรรม เป็นที่ทราบกันดีว่ามีการกลายพันธุ์ทางพันธุกรรมจำนวนมากซึ่งนำไปสู่ข้อบกพร่องใน PAH ชนิดของการกลายพันธุ์กำหนดขอบเขตที่จำกัดการสลายฟีนิลอะลานีน
PKU นั้นสืบทอดมาอย่างถดถอย ซึ่งหมายความว่าบุคคลสามารถเป็นพาหะของยีนที่เปลี่ยนแปลงไปโดยไม่ต้องพัฒนาโรค ในทำนองเดียวกันผู้ที่มีฟีนิลคีโตนูเรียสามารถให้กำเนิดบุตรที่มีสุขภาพดีได้
เฉพาะในกรณีที่พ่อแม่ทั้งสองมีการกลายพันธุ์ในองค์ประกอบทางพันธุกรรมของพวกเขาเท่านั้นที่มีความเป็นไปได้ที่ลูกหลานของพวกเขาจะพัฒนาฟีนิลคีโตนูเรีย หากทั้งพ่อและแม่ไม่เพียงแต่เป็นพาหะของยีน แต่ยังมี PKU เองด้วย เด็กทุกคนก็จะพัฒนาไปด้วยกัน
Phenylketonuria: การตรวจและการวินิจฉัย
เนื่องจากสามารถป้องกันผลร้ายแรงของฟีนิลคีโตนูเรียได้โดยเริ่มการรักษาในเวลาที่เหมาะสม การค้นพบโรคโดยเร็วที่สุดจึงเป็นสิ่งสำคัญอย่างยิ่ง ในประเทศเยอรมนี เด็ก ๆ จะได้รับการตรวจโรคประจำตัวต่างๆ รวมถึง PKU ซึ่งเป็นส่วนหนึ่งของการตรวจทั่วไป (การตรวจคัดกรองทารกแรกเกิด) ในวันที่สามหลังคลอด
แมสสเปกโตรเมทรีตีคู่
ปัจจุบันมีการวินิจฉัยความผิดปกติทางเมตาบอลิซึมที่มีมาแต่กำเนิดจำนวนมากโดยใช้สิ่งที่เรียกว่า tandem mass spectrometry ช่วยให้ตรวจเลือดของทารกแรกเกิดได้อย่างรวดเร็วและง่ายดาย นอกจากฟีนิลคีโตนูเรียแล้ว แพทย์สามารถตรวจพบโรคอื่นๆ ได้มากกว่า 20 โรคภายในไม่กี่นาที
การทดสอบ Guthrie
การทดสอบ Guthrie ซึ่งตั้งชื่อตามนักประดิษฐ์ ทำให้สามารถวินิจฉัย PKU ได้ เมื่อต้องการทำเช่นนี้ เลือดจำนวนเล็กน้อยจะถูกนำออกจากส้นเท้าของเด็กและทาลงบนกระดาษกรอง ในห้องปฏิบัติการ คุณสามารถระบุได้ว่าความเข้มข้นของฟีนิลอะลานีนเพิ่มขึ้นหรือไม่
การทดสอบ Guthrie ถูกนำมาใช้ในทศวรรษที่ 1960 และเป็นวิธีมาตรฐานในการวินิจฉัยฟีนิลคีโตนูเรียมายาวนาน อย่างไรก็ตาม มีข้อเสียเมื่อเทียบกับแมสสเปกโตรเมตรีแบบตีคู่ การทดสอบ Guthrie จะให้ผลลัพธ์หลังจากผ่านไปห้าวันเท่านั้น ซึ่งมีแนวโน้มที่จะเกิดข้อผิดพลาดเช่นกัน ตัวอย่างเช่น ปัจจัยต่างๆ เช่น อาหารของเด็กหรือการรักษาด้วยยาปฏิชีวนะที่เป็นไปได้ทำให้ผลลัพธ์เป็นเท็จ ในบางประเทศยังคงใช้การทดสอบ Guthrie แต่ในเยอรมนีมักจะไม่ใช้อีกต่อไป
การทดสอบอื่นๆ
หากการตรวจคัดกรองทารกแรกเกิดพบความสงสัยในฟีนิลคีโตนูเรีย การตรวจต่อไปเพื่อยืนยัน นอกจากนี้ยังสามารถใช้เพื่อตรวจสอบความเข้มข้นที่แน่นอนของฟีนิลอะลานีนในเลือด
สุดท้าย สิ่งสำคัญคือต้องแยกแยะว่า PKU ทั่วไป (คลาสสิก) หรือผิดปรกติหรือไม่ นอกจากนี้ยังมีการทดสอบพิเศษ เช่น การทดสอบความเครียด tetrahydrobiopterin ความแตกต่างมีความสำคัญเนื่องจากฟีนิลคีโตนูเรียที่ผิดปกติได้รับการปฏิบัติแตกต่างจากรูปแบบคลาสสิก
ตรวจน้ำคร่ำ
Phenylketonuria สามารถวินิจฉัยได้ในระหว่างตั้งครรภ์ (การวินิจฉัยก่อนคลอด) ในการทำเช่นนี้จะมีการนำน้ำคร่ำจำนวนเล็กน้อยออกจากถุงน้ำคร่ำของแม่และตรวจสอบเซลล์ของทารกในครรภ์ที่อยู่ในนั้น สามารถระบุข้อบกพร่องทางพันธุกรรมที่ทำให้เกิด PKU ได้ด้วยวิธีนี้
ด้วยการตรวจคัดกรองทารกแรกเกิด phenylketonuria มักถูกค้นพบเร็วพอที่จะรักษาได้ เนื่องจากการทดสอบน้ำคร่ำมักเกี่ยวข้องกับความเสี่ยง การนำไปใช้เพื่อวินิจฉัย PKU มักจะไม่มีประโยชน์
ฟีนิลคีโตนูเรีย: การรักษา
มีเพียงวิธีเดียวที่จะต่อต้านฟีนิลอะลานีนส่วนเกินใน PKU: เด็กที่ได้รับผลกระทบต้องปฏิบัติตามอาหารพิเศษเพื่อรับฟีนิลอะลานีนในอาหารให้น้อยที่สุด พวกเขายังต้องการผลิตภัณฑ์เสริมอาหารบางชนิดเพื่อทดแทนสารที่จะเกิดขึ้นจากฟีนิลอะลานีนในเด็กที่มีสุขภาพดี
การบำบัดจะต้องเริ่มต้นก่อนที่อาการแรกของความผิดปกติของพัฒนาการจะเกิดขึ้น นั่นคือ ภายในสองเดือนแรกของชีวิต ความเสียหายของสมองที่เกิดขึ้นแล้วไม่สามารถย้อนกลับได้
โภชนาการบำบัดด้วยอาหาร PKU
โปรตีนจากธรรมชาติทั้งหมดประกอบด้วยกรดอะมิโนฟีนิลอะลานีนประมาณห้าเปอร์เซ็นต์ อาหารส่วนใหญ่มีมากกว่าที่ร่างกายต้องการ นี่ไม่ใช่ปัญหาสำหรับคนที่มีสุขภาพดีเพราะเขาสลายและขับฟีนิลอะลานีนในปริมาณที่มากเกินไป
ในทางกลับกัน ผู้ป่วยที่เป็น phenylketonuria จำเป็นต้องทำโดยไม่มีโปรตีนจากธรรมชาติเป็นส่วนใหญ่ ผลิตภัณฑ์พิเศษที่ผลิตขึ้นเพื่ออุตสาหกรรมทดแทนส่วนประกอบอาหารที่ขาดหายไป ในอีกด้านหนึ่ง เราต้องการป้องกันไม่ให้มีฟีนิลอะลานีนมากเกินไป ในทางกลับกัน ผู้ป่วยควรได้รับสารที่อาจจะไม่เพียงพอ เช่น ไทโรซีน ซึ่งเกิดจากฟีนิลอะลานีน
อย่างไรก็ตาม เป้าหมายของอาหาร PKU ไม่ใช่การหยุดการดูดซึมฟีนิลอะลานีนอย่างสมบูรณ์ เนื่องจากร่างกายต้องการกรดอะมิโนจำนวนหนึ่งสำหรับกระบวนการเผาผลาญที่สำคัญ การมีสมาธิที่เหมาะสมถือเป็นความท้าทายที่ยิ่งใหญ่สำหรับแพทย์และผู้ป่วย และต้องมีวินัยอย่างมาก
อาหารจึงต้องปฏิบัติตามตลอดชีวิต แต่โดยเฉพาะอย่างยิ่งอย่างเคร่งครัดจนถึงอายุหกขวบ เนื่องจากสมองมีการพัฒนาอย่างมากจนถึงวัยนี้ ดังนั้นจึงมีความอ่อนไหวต่อความเสียหายเป็นพิเศษ ในวัยผู้ใหญ่ฟีนิลอะลานีนที่มีความเข้มข้นสูงจะไม่ทำให้สมองเสียหายเหมือนที่ทำในเด็ก อย่างไรก็ตาม สิ่งเหล่านี้สามารถเป็นตัวกระตุ้นให้เกิดการร้องเรียนทางระบบประสาทอื่นๆ เช่น สมาธิไม่ดีหรือปฏิกิริยาช้าลง
การรักษาอาหารสำหรับฟีนิลคีโตนูเรียควรเริ่มต้นที่ศูนย์โรคเมตาบอลิผู้เชี่ยวชาญ เพราะทุกคลินิกไม่สามารถสั่งสอนผู้ปกครองเรื่องอาหารได้ วิธีนี้ทำได้ดีที่สุดด้วยความช่วยเหลือจากการให้คำปรึกษาเรื่องอาหาร ซึ่งจะแสดงวิธีการควบคุมอาหารและตรวจสอบระดับฟีนิลอะลานีนในเลือดเป็นประจำ
การรักษาฟีนิลคีโตนูเรียผิดปรกติ
ในรูปแบบที่ผิดปกติของ PKU โคเอ็นไซม์ BH4 ที่หายไปและสารส่งสารบางอย่างเช่นโดปามีนและเซโรโทนินจะถูกแทนที่เทียม ในบางกรณี ผู้ป่วยต้องรับประทานอาหารที่มีฟีนิลอะลานีนต่ำด้วย
Phenylketonuria ระหว่างตั้งครรภ์
หากคุณกำลังตั้งครรภ์และมีฟีนิลคีโตนูเรียด้วยตัวเอง คุณควรพิจารณาสิ่งต่อไปนี้:
- ยึดมั่นในอาหารของคุณโดยเฉพาะอย่างยิ่งอย่างเคร่งครัด!
- ตรวจค่าเลือดฟีนิลอะลานีนเป็นระยะๆ เนื่องจากความเข้มข้นของฟีนิลอะลานีนในเด็กที่ยังไม่เกิดนั้นสูงเป็นสองเท่าของในหญิงตั้งครรภ์ นอกจากสมองแล้ว ยังทำลายหัวใจและดวงตาของทารกในครรภ์ได้อีกด้วย ความผิดปกติของโครงกระดูกของเด็กอาจเกิดขึ้นได้จากระดับฟีนิลอะลานีนที่สูงในระหว่างตั้งครรภ์
- เพื่อแยกแยะความเสียหายของสมองต่อเด็กในการตั้งครรภ์ระยะแรก ผู้หญิงที่เป็นโรคฟีนิลคีโตนูเรียควรวางแผนการตั้งครรภ์อย่างระมัดระวังและระมัดระวังตั้งแต่เริ่มแรก
- ในหญิงตั้งครรภ์ที่เป็นโรคฟีนิลคีโตนูเรีย การรับประทานอาหารที่ไม่ปฏิบัติตามอย่างเคร่งครัดสามารถนำไปสู่การแท้งบุตรได้ (การทำแท้งโดยธรรมชาติ)
- ไม่ว่าในกรณีใดสตรีมีครรภ์ที่มีฟีนิลคีโตนูเรียควรขอคำแนะนำและคำแนะนำจากแพทย์
Phenylketonuria: โรคและการพยากรณ์โรค
หากตรวจพบฟีนิลคีโตนูเรียตั้งแต่เนิ่นๆ หากเป็นไปได้ในทารกแรกเกิด และหากปฏิบัติตามอาหารพิเศษของ PKU การพยากรณ์โรคมักจะดี เด็กมีพัฒนาการทางจิตวิญญาณและมีอายุขัยเฉลี่ย
อย่างไรก็ตาม หากปล่อยทิ้งไว้โดยไม่รักษา ความเสียหายของสมองจะนำไปสู่ความผิดปกติทางพัฒนาการทางจิตอย่างรุนแรงซึ่งไม่สามารถแก้ไขได้ในภายหลัง ผู้ที่ได้รับผลกระทบมีอายุขัยเฉลี่ย แต่ความฉลาดทางสติปัญญามักจะต่ำกว่าเกณฑ์ปกติเกือบตลอดเวลา
phenylketonuria รูปแบบผิดปกติที่หายากซึ่งมีการขาด BH4 เป็นกรณีพิเศษ ฟีนิลคีโตนูเรียที่แปรผันนี้สามารถนำไปสู่ความเสียหายทางระบบประสาทที่ก้าวหน้าและเป็นตะคริวอย่างรุนแรงแม้จะรับประทานอาหาร
แท็ก: การดูแลทันตกรรม การป้องกัน ยาสมุนไพร ยาสามัญประจำบ้าน
















.jpg)